Hello,
I’m a newbie to Psi4, and indeed, to the field of computational chemistry. I believe in free (as in speech, not only as in beer) software, and a unfortunate thing I perceived when starting my graduate studies is that the field is still dominated by proprietary tools. So, first and foremost, I’d like to praise the Psi4 developers for the job you’re doing for science and all of us.
To learn Psi4, I started with the simplest possible system. A diatomic molecule. I wrote a small python script(PsiAPI) trying to plot potential energy versus bond length, first for hydrogen, and then for chlorine, using four different functionals (hf, scf, b3lyp and mp2) and four different basis sets (sto-3g, 3-21G, 6-311G and 6-311G(d,p)) combined:
'''
Plots E vs. d for diatomic chlorine. To change for hydrogen, you can replace
both Cl's in geometry section for H, change range(150) to range(50)
and internuclearD = 1.5 + n/100 to internuclearD = 0.5 + n/100.
'''
import psi4
import time
psi4.set_memory('4GB')
for method in ['hf', 'scf', 'b3lyp', 'mp2']:
for base in ['sto-3g', '3-21G', '6-311G', '6-311G(d,p)']:
for n in range(150):
internuclearD = 1.5 + n/100
#psi4.core.set_output_file('curvaPotencialCl2_' + str(n) + '.dat', False)
complexo = psi4.geometry('''
0 1
Cl 0.00000 0.00000 0.00000
Cl 0.00000 0.00000 ''' + str(internuclearD))
try:
t1 = time.time()
energia = psi4.energy(method + '/' + base, molecule = complexo)
t2 = time.time()
print('resultado;' + str(method) + ';' + str(base) + ';' + str(internuclearD) + ';' + str(energia) + ';' + str(t2 - t1))
except:
print('resultado;' + str(method) + ';' + str(base) + ';;')For hydrogen the results were just as expected. Energy varied a bit depending on method used, but all had the expected shape, a continuous curve with a minimum arond 0.7 angström:
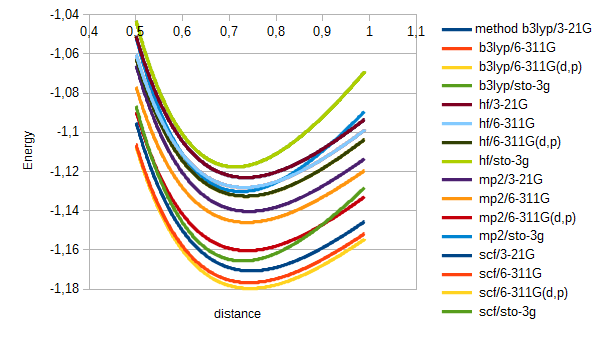
For chlorine I got a strange result, for all functionals except b3lyp. When ploting the data, after de minimum the curve loses continuity, splitting in two, with large oscilations in energy between consecutive distance values.
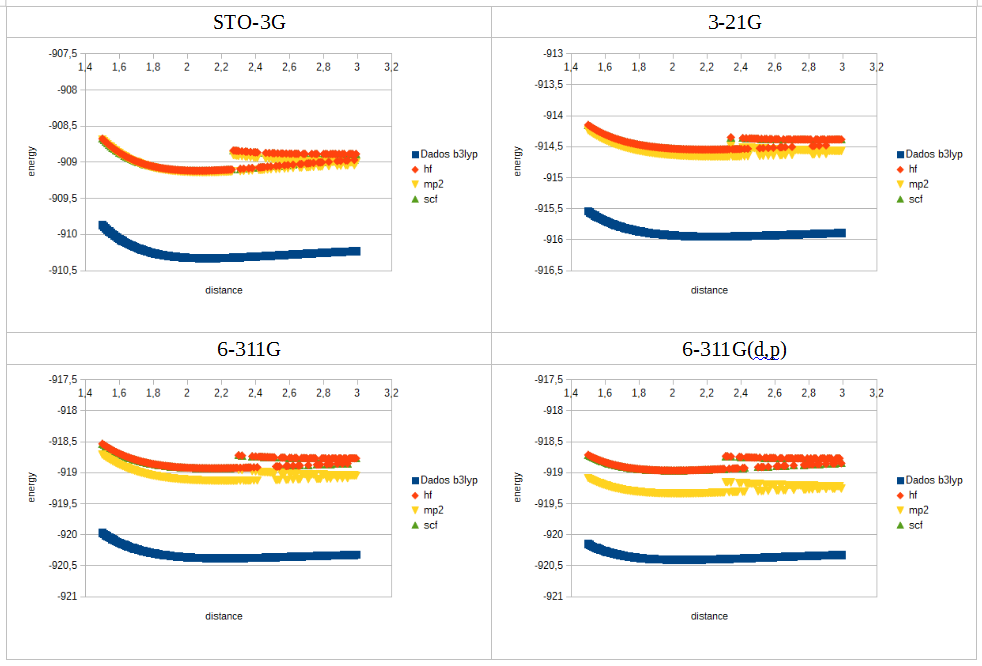
What does cause this odd behaviour?