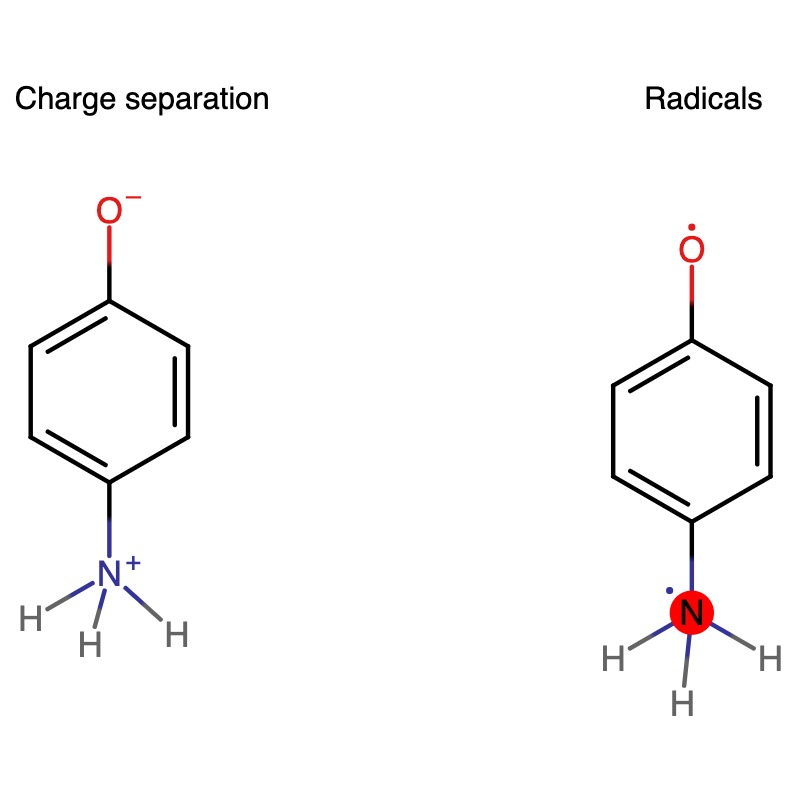
I was wondering about how to specify formal charges in molecules, particularly zwitterions. When I create a molecule using an xyz-file (see attached), this charge separation isn’t included since the molecule’s total charge is zero and only atom positions are encoded in the file. To my understanding, this means that Psi4 interprets this as two radicals (see attached image). Or am I off track since formal charges (as the name suggests) are a formality, and I should let Psi4 figure out the physical charge distribution from the atom positions and molecular charge alone?
The right option here is indeed to let Psi figure out the physical charge distribution from your SCF.
Here is an example of using Psi to compute several properties, including Lowdin and Mulliken charges. You can use those to sanity-check Psi’s solution, if you wish. You can also use the OEProp object if you want a function that you can feed a wavefunction into. Be warned that this will work for SCF, but is trickier for more general wavefunctions. (This has to do with how you define the density for correlated wavefunctions, and I do not believe it is relevant to your question.)