Dear colleagues,
I wrote a simple python script (archive psi4teste.py) to run 2 jobs in a PBS queue in a cluster (psi41.pbs and psi42.pbs files) to calculate the SAPT energy (sapt2+(3)dmp2/aug-cc-pVTZ level) of 2 files (metano1_1.dat and metano1_2.dat files).
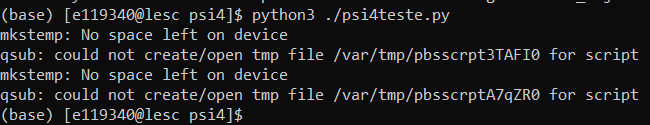
However, the energy calculations crash and the output files are incomplete (metano1_1.out and metano1_2.out files), giving the error below:
Traceback (most recent call last):
File “/ home / sw / anaconda3 / envs / p4env / bin / psi4”, line 287, in
exec (content)
File “”, line 33, in
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/driver.py”, line 556, in energy
wfn = procedures [‘energy’] [lowername] (lowername, molecule = molecule, ** kwargs)
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/procrouting/proc.py”, line 3309, in run_sapt
dimer_wfn = scf_helper (‘RHF’, molecule = sapt_dimer, ** kwargs)
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/procrouting/proc.py”, line 1363, in scf_helper
e_scf = scf_wfn.compute_energy ()
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/procrouting/scf_proc/scf_iterator.py”, line 84, in scf_compute_energy
self.initialize ()
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/procrouting/scf_proc/scf_iterator.py”, line 198, in scf_initialize
self.initialize_jk (self.memory_jk_, jk = jk)
File “/home/sw/anaconda3/envs/p4env/lib//python3.7/site-packages/psi4/driver/procrouting/scf_proc/scf_iterator.py”, line 125, in initialize_jk
jk.initialize ()
RuntimeError:
Fatal Error: PSIO Error
Error occurred in file: /scratch/psilocaluser/conda-builds/psi4-multiout_1557940846948/work/psi4/src/psi4/libpsio/error.cc online: 128
The most recent 5 function calls were:
psi::PSIO::rw(unsigned long, char*, psi::psio_address, unsigned long, int)
psi::PSIO::write_entry(unsigned long, char const*, char*, unsigned long)
psi::DiskDFJK::initialize_JK_core()
Any suggestions as to what this error might be?
All the files i mentioned are available in this link:
https://drive.google.com/drive/folders/184LIMsbsYQkGD6mpyMH9qgIix74GsQIf?usp=sharing
Sorry for my lack of experience.
Thanks in advance for your time!