molecule {
0 1
O 0.000000 0.000000 6.000000
H 0.000000 1.431500 4.890600
H 0.000000 -1.431500 4.890600
–
0 2
O 0.000000 0.000000 0.000000
O 0.000000 2.503900 0.000000
H 0.000000 -0.424700 -1.839500
units bohr
symmetry c1
no_reorient
no_com
}
set {
reference uhf
scf_type df
basis cc-pVDZ
}
energy(‘sapt0’)
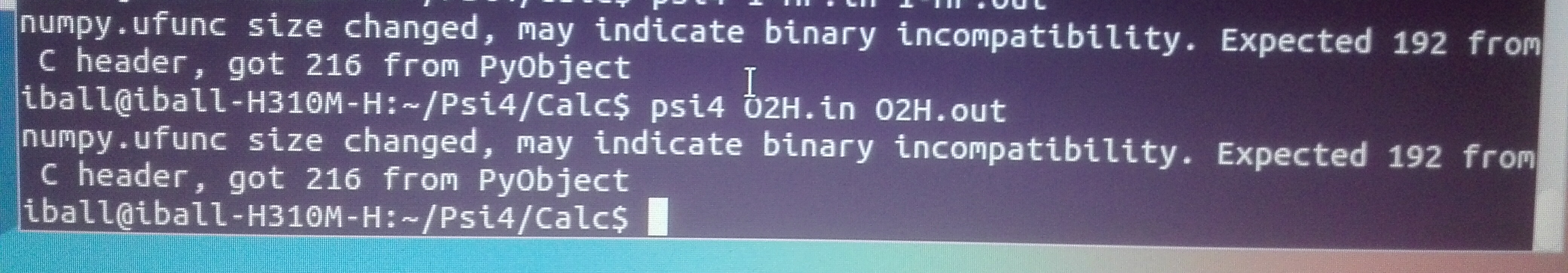
i have tried to run the above input file in psi4 for sapt analysis which i have got psi4 sample file. unfortunately, the ‘out’ file was showing error something like following…
What version of Psi4 are you using? Your traceback leads me to suspect the version you are using is rather old. Depending on how old it is, open-shell SAPT0 may not exist in the code.
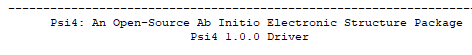
The information I’m looking for will be in the first six lines of the output file. If you’re using the latest official release, it should look something like
-----------------------------------------------------------------------
Psi4: An Open-Source Ab Initio Electronic Structure Package
Psi4 1.3 release
Git: Rev {HEAD} 20e5c7e
Just as I thought, you need to update your version of Psi. Upgrade to Psi 1.3, and you can do open-shell SAPT0. Get the latest version here. Let me know if that works!
Where did you find a link to that version of Psi? Something that old shouldn’t be on this site.
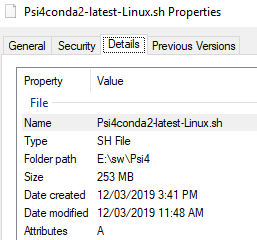
I don’t remember the version that I have downloaded but I am sure it was from this site psicode.org.
Thanks for your consistent reply and support for my each queries…
so, I’ll download the Psi4 1.3 version from the your link or the site.