Hi all,
Edit: Forgot to add the s/w version:
Psi4: An Open-Source Ab Initio Electronic Structure Package Psi4 1.9.1 release Git: Rev {} zzzzzzz
I’m having a look at molecules similar to the one below (paper: https://pubs.acs.org/doi/epdf/10.1021/jm501853m?ref=article_openPDF) and trying to determine what kind of stabilising interaction is occurring here (the paper just notes energetic stability with NBO-style analysis).
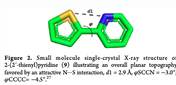
The molecule I’m working with is similar (Me added) but the orientation is identical. At that distance, there should be no meaningful orbital overlap and applying topology analysis (AIM) confirm this with no BCP found between the atoms. RDG and similar analyses find weak (vdW) interactions but that doesn’t tell me much.
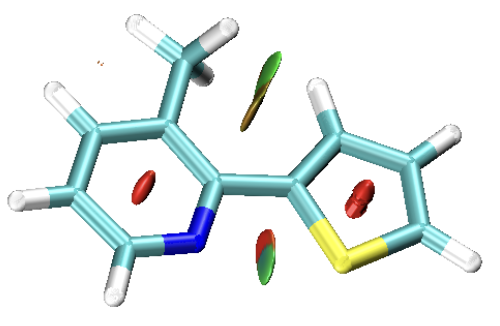
I suspect a very weak, purely electrostatic interaction with the N-S but I’d like to confirm this.
I don’t have other s/w to run IQA or EDA so I thought running an I-SAPT might do the job. I fragmented the system with the following scheme, partitioning it into the N atom, the S atom and the rest as the linker:
mol = psi4.core.Molecule.from_arrays(
elez=[7, 16, 6, 6, 6, 6, 6, 6, 6, 6, 6, 6, 1, 1, 1, 1, 1, 1, 1, 1, 1],
fragment_separators=[1, 2],
fix_com=True,
fix_orientation=True,
fix_symmetry='c1',
fragment_multiplicities=[4, 3, 2],
molecular_charge=0,
molecular_multiplicity=1,
geom= [
-1.042343, 1.158938, 0.499259,
1.696223, 1.399517, -0.069361,
-1.081346, -2.554510, -0.320227,
-1.493733, -1.167502, 0.083810,
-0.613929, -0.060883, 0.126590,
-2.308266, 1.348321, 0.835267,
-3.248811, 0.326651, 0.824373,
-2.819456, -0.934346, 0.443143,
0.812693, -0.097073, -0.212125,
1.654216, -1.101341, -0.641313,
2.987438, -0.670680, -0.850246,
3.153836, 0.657597, -0.579786,
-1.930442, -3.234338, -0.267764,
-0.698269, -2.577692, -1.342006,
-0.298922, -2.946431, 0.332039,
-2.582937, 2.356559, 1.124764,
-4.276064, 0.513314, 1.104102,
-3.520445, -1.759684, 0.421612,
1.349723, -2.118268, -0.808468,
3.781612, -1.321003, -1.186089,
4.060473, 1.235711, -0.655552
])
And ran it with the following options:
psi4.set_options({
"basis": 'def2-TZVP',
"scf_type": "df",
"guess": "sad",
"freeze_core": True,
"fisapt_do_plot": True, # For extra analysis
# "stability_analysis": 'follow'
})
psi4.set_num_threads(32)
psi4.set_memory('16000 MB')
energ_sp, wfn_sp = psi4.energy('fisapt0', molecule=mol, return_wfn=True)
This system passed the multiplicity and sanity checks and ran okay but the energies are ludicrous, so obviously there’s something wrong in the setup.
Electrostatics -9938.78880106 [mEh] -6236.68413054 [kcal/mol] -26094.28640217 [kJ/mol]
Elst10,r -9938.78880106 [mEh] -6236.68413054 [kcal/mol] -26094.28640217 [kJ/mol]
Exchange 13349.38739217 [mEh] 8376.86705772 [kcal/mol] 35048.81176948 [kJ/mol]
Exch10 13349.38739217 [mEh] 8376.86705772 [kcal/mol] 35048.81176948 [kJ/mol]
Exch10(S^2) 7521.06628052 [mEh] 4719.54034394 [kcal/mol] 19746.55679903 [kJ/mol]
Induction -2553.50080330 [mEh] -1602.34594537 [kcal/mol] -6704.21543543 [kJ/mol]
Ind20,r -75641.38274992 [mEh] -47465.68428521 [kcal/mol] -198596.42304932 [kJ/mol]
Exch-Ind20,r 40446.12877136 [mEh] 25380.32898166 [kcal/mol] 106191.29645926 [kJ/mol]
delta HF,r (2) 32641.75317526 [mEh] 20483.00935818 [kcal/mol] 85700.91115463 [kJ/mol]
Induction (A<-B) -1734.75125958 [mEh] -1088.57285004 [kcal/mol] -4554.58880455 [kJ/mol]
Induction (B<-A) -818.74954372 [mEh] -513.77309533 [kcal/mol] -2149.62663088 [kJ/mol]
Dispersion 3099.23475684 [mEh] 1944.79917138 [kcal/mol] 8137.03973304 [kJ/mol]
Disp20 -452.34656085 [mEh] -283.85175237 [kcal/mol] -1187.63573190 [kJ/mol]
Exch-Disp20 3551.58131769 [mEh] 2228.65092374 [kcal/mol] 9324.67546494 [kJ/mol]
From searching the forum, I know atomic fragments is recommended against but as you can see, with this system, bigger fragments would be cutting pi-bonds and I’m guessing that’s just as problematic. If I make the rings themselves fragments, there doesn’t seem to be the 3rd ‘linker’ fragment any more.
Anyone got a more appropriate fragment scheme, advice for what else I’m missing or recommendation for a method I should use instead? I saw mention of A-SAPT but this doesn’t appear to be implemented. Cheers all.
EDIT: Just noticed that I used the wrong basis set, should be jun-cc-pvdz. However, I get the following error:
RuntimeError:
Fatal Error: Monomer B: A Matrix is not SPD
I’ve seen a fix for this (“stability_analysis”: ‘follow’) but as I’m doing RHF, it’s not an option. Clues from here?