I am unable to install PSI4 in native Windows for SAPT analysis. Please help.
Could you kindly share how you are trying to install psi4 and what error you are encountering?
Yes, unfortunately the download button does not work on all browsers. Try right-clicking and selecting “Save as” or use the provided curl command.
Psiconda is downloaded but in each try it wants permit to install. And is not working properly. Will you help?
I want to start a SAPT calculation on a hydrogen bonded system. How can I stat with the input system.
Partha
I draw the input but the calculation is failed. Please help showing the distribution of different kinds of energy.
Partha
Python 3.8.16 | packaged by conda-forge | (default, Feb 1 2023, 15:53:35) [MSC v.1929 64 bit (AMD64)] on win32
Type “help”, “copyright”, “credits” or “license” for more information.
Example SAPT computation for pyrazine (i.e., pyrazine).
pyrazine
dimer = pyrazinne.geometry(“”"
… 0 1
… … 7 -0.500264000 -0.463306000 0.704771000
… … 7 0.722836000 0.571897000 3.010784000
… … 6 -0.385290000 -1.178101000 1.825677000
… … 6 -0.000940000 0.775280000 0.741348000
… … 6 0.606909000 1.284810000 1.888933000
… … 6 0.221047000 -0.663570000 2.970375000
… … 1 -0.787134000 -2.187994000 1.812223000
… … 1 -0.096719000 1.360597000 -0.171101000
… … 1 1.009532000 2.293997000 1.902708000
… … 1 0.304265000 -1.260508000 3.874617000
… … –
… … 0 1
… …
… … 7 -0.872739000 0.366051000 -2.302687000
… … 1 -1.572821000 0.410798000 -3.036553000
… … 1 -1.233980000 -0.238213000 -1.567701000
… … 6 0.383159000 -0.173410000 -2.816852000
… … 1 1.117308000 -0.204707000 -2.003447000
… … 1 0.774605000 0.493810000 -3.590949000
… … 1 0.310225000 -1.187430000 -3.242146000
… … units angstrom
… … “”“)
Traceback (most recent call last):
File “”, line 1, in
NameError: name ‘pyrazinne’ is not defined
0 1
File “”, line 1
0 1
^
SyntaxError: invalid syntax
… 7 -0.500264000 -0.463306000 0.704771000
File “”, line 1
… 7 -0.500264000 -0.463306000 0.704771000
^
SyntaxError: invalid syntax
… 7 0.722836000 0.571897000 3.010784000
File “”, line 1
… 7 0.722836000 0.571897000 3.010784000
^
SyntaxError: invalid syntax
… 6 -0.385290000 -1.178101000 1.825677000
File “”, line 1
… 6 -0.385290000 -1.178101000 1.825677000
^
SyntaxError: invalid syntax
… 6 -0.000940000 0.775280000 0.741348000
File “”, line 1
… 6 -0.000940000 0.775280000 0.741348000
^
SyntaxError: invalid syntax
… 6 0.606909000 1.284810000 1.888933000
File “”, line 1
… 6 0.606909000 1.284810000 1.888933000
^
SyntaxError: invalid syntax
… 6 0.221047000 -0.663570000 2.970375000
File “”, line 1
… 6 0.221047000 -0.663570000 2.970375000
^
SyntaxError: invalid syntax
… 1 -0.787134000 -2.187994000 1.812223000
File “”, line 1
… 1 -0.787134000 -2.187994000 1.812223000
^
SyntaxError: invalid syntax
… 1 -0.096719000 1.360597000 -0.171101000
File “”, line 1
… 1 -0.096719000 1.360597000 -0.171101000
^
SyntaxError: invalid syntax
… 1 1.009532000 2.293997000 1.902708000
File “”, line 1
… 1 1.009532000 2.293997000 1.902708000
^
SyntaxError: invalid syntax
… 1 0.304265000 -1.260508000 3.874617000
File “”, line 1
… 1 0.304265000 -1.260508000 3.874617000
^
SyntaxError: invalid syntax
… –
File “”, line 1
… –
^
SyntaxError: invalid syntax
… 0 1
File “”, line 1
… 0 1
^
SyntaxError: invalid syntax
…
Ellipsis
… 7 -0.872739000 0.366051000 -2.302687000
File “”, line 1
… 7 -0.872739000 0.366051000 -2.302687000
^
SyntaxError: invalid syntax
… 1 -1.572821000 0.410798000 -3.036553000
File “”, line 1
… 1 -1.572821000 0.410798000 -3.036553000
^
SyntaxError: invalid syntax
… 1 -1.233980000 -0.238213000 -1.567701000
File “”, line 1
… 1 -1.233980000 -0.238213000 -1.567701000
^
SyntaxError: invalid syntax
… 6 0.383159000 -0.173410000 -2.816852000
File “”, line 1
… 6 0.383159000 -0.173410000 -2.816852000
^
SyntaxError: invalid syntax
… 1 1.117308000 -0.204707000 -2.003447000
File “”, line 1
… 1 1.117308000 -0.204707000 -2.003447000
^
SyntaxError: invalid syntax
… 1 0.774605000 0.493810000 -3.590949000
File “”, line 1
… 1 0.774605000 0.493810000 -3.590949000
^
SyntaxError: invalid syntax
… 1 0.310225000 -1.187430000 -3.242146000
File “”, line 1
… 1 0.310225000 -1.187430000 -3.242146000
^
SyntaxError: invalid syntax
… units angstrom
File “”, line 1
… units angstrom
^
SyntaxError: invalid syntax
… “””)
… 0 1
… … 7 -0.500264000 -0.463306000 0.704771000
… … 7 0.722836000 0.571897000 3.010784000
… … 6 -0.385290000 -1.178101000 1.825677000
… … 6 -0.000940000 0.775280000 0.741348000
… … 6 0.606909000 1.284810000 1.888933000
… … 6 0.221047000 -0.663570000 2.970375000
… … 1 -0.787134000 -2.187994000 1.812223000
… … 1 -0.096719000 1.360597000 -0.171101000
… … 1 1.009532000 2.293997000 1.902708000
… … 1 0.304265000 -1.260508000 3.874617000
… … –
… … 0 1
… …
… … 7 -0.872739000 0.366051000 -2.302687000
… … 1 -1.572821000 0.410798000 -3.036553000
… … 1 -1.233980000 -0.238213000 -1.567701000
… … 6 0.383159000 -0.173410000 -2.816852000
… … 1 1.117308000 -0.204707000 -2.003447000
… … 1 0.774605000 0.493810000 -3.590949000
… … 1 0.310225000 -1.187430000 -3.242146000
… … units angstrom
… … “”“)
File “”, line 24
… “””)
0 1
… 7 -0.500264000 -0.463306000 0.704771000
… 7 0.722836000 0.571897000 3.010784000
… 6 -0.385290000 -1.178101000 1.825677000
… 6 -0.000940000 0.775280000 0.741348000
… 6 0.606909000 1.284810000 1.888933000
… 6 0.221047000 -0.663570000 2.970375000
… 1 -0.787134000 -2.187994000 1.812223000
… 1 -0.096719000 1.360597000 -0.171101000
… 1 1.009532000 2.293997000 1.902708000
… 1 0.304265000 -1.260508000 3.874617000
… –
… 0 1
…
… 7 -0.872739000 0.366051000 -2.302687000
… 1 -1.572821000 0.410798000 -3.036553000
… 1 -1.233980000 -0.238213000 -1.567701000
… 6 0.383159000 -0.173410000 -2.816852000
… 1 1.117308000 -0.204707000 -2.003447000
… 1 0.774605000 0.493810000 -3.590949000
… 1 0.310225000 -1.187430000 -3.242146000
… units angstrom
… “”")
^
SyntaxError: invalid syntax
The syntax for your input geometry is wrong.
It should look like, for example
dimer = psi4.geometry("""
0 1
C 0.000000 -0.667578 -2.124659
C 0.000000 0.667578 -2.124659
H 0.923621 -1.232253 -2.126185
H -0.923621 -1.232253 -2.126185
H -0.923621 1.232253 -2.126185
H 0.923621 1.232253 -2.126185
--
0 1
C 0.000000 0.000000 2.900503
C 0.000000 0.000000 1.693240
H 0.000000 0.000000 0.627352
H 0.000000 0.000000 3.963929
units angstrom
""")
You can see an example SAPT calculation here: PsiAPI Tutorial: Using Psi4 as a Python Module
The syntax error is found
Python 3.8.16 | packaged by conda-forge | (default, Feb 1 2023, 15:53:35) [MSC v.1929 64 bit (AMD64)] on win32
Type “help”, “copyright”, “credits” or “license” for more information.
Example SAPT computation for pyrazine (i.e., pyrazine).
pyrazine
dimer = pyrazine.geometry(“”"
…
… 0 1
… N -0.500264000 -0.463306000 0.704771000
… N 0.722836000 0.571897000 3.010784000
… C -0.385290000 -1.178101000 1.825677000
… C -0.000940000 0.775280000 0.741348000
… C 0.606909000 1.284810000 1.888933000
… C 0.221047000 -0.663570000 2.970375000
… H -0.787134000 -2.187994000 1.812223000
… H -0.096719000 1.360597000 -0.171101000
… H 1.009532000 2.293997000 1.902708000
… H 0.304265000 -1.260508000 3.874617000
… –
… 0 1
…
… N -0.872739000 0.366051000 -2.302687000
… H -1.572821000 0.410798000 -3.036553000
… H -1.233980000 -0.238213000 -1.567701000
… C 0.383159000 -0.173410000 -2.816852000
… H 1.117308000 -0.204707000 -2.003447000
… H 0.774605000 0.493810000 -3.590949000
… h 0.310225000 -1.187430000 -3.242146000
… units angstrom
… “”")
Traceback (most recent call last):
File “”, line 1, in
NameError: name ‘pyrazine’ is not defined
Partha
Sir, i input this for SAPT analysis# SAPT computationfor pyrazinemethyl amine (i.e., pyrazinemethyl amine). # Test dimer =psi4.geometry(“”“0 1N -0.500264000 -0.463306000 0.704771000N 0.722836000 0.571897000 3.010784000C -0.385290000 -1.178101000 1.825677000C -0.000940000 0.775280000 0.741348000C 0.606909000 1.284810000 1.888933000C 0.221047000 -0.663570000 2.970375000H -0.787134000 -2.187994000 1.812223000H -0.096719000 1.360597000 -0.171101000H 1.009532000 2.293997000 1.902708000H 0.304265000 -1.260508000 3.874617000–0 1 N -0.872739000 0.366051000 -2.302687000H -1.572821000 0.410798000 -3.036553000H -1.233980000 -0.238213000 -1.567701000 C 0.383159000 -0.173410000 -2.816852000H 1.117308000 -0.204707000 -2.003447000H 0.774605000 0.493810000 -3.590949000H 0.310225000 -1.187430000 -3.242146000units angstrom”“”)
psi4.set_options({‘scf_type’: ‘df’, ‘freeze_core’: True})psi4.energy(‘sapt0/jun-cc-pvdz’, molecule=dimer)But the result isPython 3.8.16 | packaged by conda-forge | (default,Feb 1 2023, 15:53:35) [MSC v.1929 64 bit(AMD64)] on win32Type “help”, “copyright”,“credits” or “license” for more information
.>>> impot psi4 File"“, line 1 impot psi4
^SyntaxError: invalid syntax>>> import psi4>>> # SAPT computation for pyrazinemethyl amine (i.e., pyrazinemethylamine).>>> # Test dimer = psi4.geometry(”“”>>> 0 1 File"“, line 1 0 1 ^SyntaxError: invalid syntax>>> N -0.500264000 -0.463306000 0.704771000
File”“, line 1 N -0.500264000 -0.463306000 0.704771000 ^SyntaxError: invalid syntax>>> N 0.722836000 0.571897000 3.010784000 File”“, line 1 N 0.722836000 0.571897000 3.010784000 ^SyntaxError: invalid syntax>>> C -0.385290000 -1.178101000 1.825677000 File”“, line 1 C -0.385290000 -1.178101000 1.825677000 ^SyntaxError: invalid syntax>>> C -0.000940000 0.775280000 0.741348000 File”“, line 1 C -0.000940000 0.775280000 0.741348000 ^SyntaxError: invalid syntax>>> C 0.606909000 1.284810000 1.888933000 File”“, line 1 C 0.606909000 1.284810000 1.888933000 ^SyntaxError: invalid syntax>>> C 0.221047000 -0.663570000 2.970375000 File”“, line 1 C 0.221047000 -0.663570000 2.970375000 ^SyntaxError: invalid syntax>>> H -0.787134000 -2.187994000 1.812223000 File”“, line 1 H -0.787134000 -2.187994000 1.812223000 ^SyntaxError: invalid syntax>>> H -0.096719000 1.360597000 -0.171101000 File”“, line 1 H -0.096719000 1.360597000 -0.171101000 ^SyntaxError: invalid syntax>>> H 1.009532000 2.293997000 1.902708000 File”“, line 1 H 1.009532000 2.293997000 1.902708000 ^SyntaxError: invalid syntax>>> H 0.304265000 -1.260508000 3.874617000 File”“, line 1 H 0.304265000 -1.260508000 3.874617000 ^SyntaxError: invalid syntax>>> – File”“, line 1 – ^SyntaxError: invalid syntax>>> 0 1 File”“, line 1 0 1 ^SyntaxError: invalid syntax>>> >>> N -0.872739000 0.366051000 -2.302687000 File”“, line 1 N -0.872739000 0.366051000 -2.302687000 ^SyntaxError: invalid syntax>>> H -1.572821000 0.410798000 -3.036553000 File”“, line 1 H -1.572821000 0.410798000 -3.036553000 ^SyntaxError: invalid syntax>>> H -1.233980000 -0.238213000 -1.567701000Traceback (most recent call last): File”“, line 1, in NameError: name ‘H’ is not defined>>> C 0.383159000 -0.173410000 -2.816852000 File”“, line 1 C 0.383159000 -0.173410000 -2.816852000 ^SyntaxError: invalid syntax>>> H 1.117308000 -0.204707000 -2.003447000 File”“, line 1 H 1.117308000 -0.204707000 -2.003447000 ^SyntaxError: invalid syntax>>> H 0.774605000 0.493810000 -3.590949000 File”“, line 1 H 0.774605000 0.493810000 -3.590949000 ^SyntaxError: invalid syntax>>> H 0.310225000 -1.187430000 -3.242146000 File”“, line 1 H 0.310225000 -1.187430000 -3.242146000 ^SyntaxError: invalid syntax>>> units angstrom File”“, line 1 units angstrom ^SyntaxError: invalid syntax>>> “””)… psi4.set_options({‘scf_type’: ‘df’, ‘freeze_core’:True})… psi4.energy(‘sapt0/jun-cc-pvdz’, molecule=dimer)will you help me correcting the input file.Partha
Python needs """ for the quotes, not “”“
Also, it looks to me that you are running this in a python interactive shell. Instead, save the input to a file and run that file with python .
Sir, You are kind enough. Will you help me to correct this input file I use pbepbe method and 6-311+(d) basis set.
SAPT computation for pyrazinemethyl amine (i.e., pyrazinemethyl amine).
dimer = psi4.geometry(“”"’’
0 1
N -0.500264000 -0.463306000 0.704771000
N 0.722836000 0.571897000 3.010784000
C -0.385290000 -1.178101000 1.825677000
C -0.000940000 0.775280000 0.741348000
C 0.606909000 1.284810000 1.888933000
C 0.221047000 -0.663570000 2.970375000
H -0.787134000 -2.187994000 1.812223000
H -0.096719000 1.360597000 -0.171101000
H 1.009532000 2.293997000 1.902708000
H 0.304265000 -1.260508000 3.874617000
0 1
N -0.872739000 0.366051000 -2.302687000
H -1.572821000 0.410798000 -3.036553000
H -1.233980000 -0.238213000 -1.567701000
C 0.383159000 -0.173410000 -2.816852000
H 1.117308000 -0.204707000 -2.003447000
H 0.774605000 0.493810000 -3.590949000
H 0.310225000 -1.187430000 -3.242146000
units angstrom
“”")
psi4.set_options({‘scf_type’: ‘df’, ‘freeze_core’: True})
psi4.energy(‘sapt0/jun-cc-pvdz’, molecule=dimer)
the system says multiplicity 1 is invalid. But I calculate a lot of DFT with charge 0 with multiplicity 1 for the system.
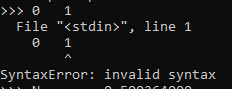
Sir, This is the input
SAPT computation for pyrazinemethyl amine (i.e., pyrazinemethyl amine).
dimer = psi4.geometry(“”"
0 1
N -0.500264000 -0.463306000 0.704771000
N 0.722836000 0.571897000 3.010784000
C -0.385290000 -1.178101000 1.825677000
C -0.000940000 0.775280000 0.741348000
C 0.606909000 1.284810000 1.888933000
C 0.221047000 -0.663570000 2.970375000
H -0.787134000 -2.187994000 1.812223000
H -0.096719000 1.360597000 -0.171101000
H 1.009532000 2.293997000 1.902708000
H 0.304265000 -1.260508000 3.874617000
0 1
N -0.872739000 0.366051000 -2.302687000
H -1.572821000 0.410798000 -3.036553000
H -1.233980000 -0.238213000 -1.567701000
C 0.383159000 -0.173410000 -2.816852000
H 1.117308000 -0.204707000 -2.003447000
H 0.774605000 0.493810000 -3.590949000
H 0.310225000 -1.187430000 -3.242146000
units angstrom
“”")
psi4.set_options({‘scf_type’: ‘df’, ‘freeze_core’: True})
psi4.energy(‘sapt0/jun-cc-pvdz’, molecule=dimer)
but there is an error
please correct the input. I tried several times. I know you are very busy but please do the correction in the input…
Yours sincerely,
Partha
See our advice on asking questions for what we expect from questions. In particular, we need a Psi4 version number and the input file enclosed in backticks.
I suspect that you have the dimer = psi4.geometry line commented out in your input file, but I can’t know that for sure due to the formatting issues.