I am trying to run a simple scan of a interatomic distance on a very simple system to locate a transition state for the reaction OH- + CH3Br → Br- + CH3OH.
I am using the following code:
memory 4 gb
molecule TS_tst{
-1 1
O 0.0000 0.0000 0.0000
H -0.2903 0.6470 0.6470
C 4.2813 0.1586 -0.7868
Br 6.2658 0.1586 -0.7868
H 3.9180 0.8853 -0.0602
H 3.9179 -0.8341 -0.5209
H 3.9179 0.4246 -1.7795
}
set {
basis 6-31G
g_convergence gau
opt_type min
}
freeze_list = """
1 xyz
3 xyz
"""
distance = 4.35
steps = 100
variation = -0.0305
counter = 0
while counter < steps:
counter = counter + 1
distance = distance + variation
distance_string = "1 3 %f" %distance
set optking fixed_distance = $distance_string
set optking frozen_cartesian $freeze_list
e, wfn = energy('b3lyp',return_wfn=True)
print("Step: %d Distance: %f and Energy: %f" %(counter,distance,e))
but I get the following error:
!----------------------------------------------------------------------------------!
! !
! Fatal Error: FIXED_DISTANCE is not a valid option. !
! Error occurred in file: /scratch/psilocaluser/conda- !
! builds/psi4-multiout_1670980379431/work/psi4/src/psi4/liboptions/liboptions. !
! h on line: 55 !
! The most recent 5 function calls were: !
! psi::Options::use(std::__cxx11::basic_string<char, std::char_traits<char>, !
! std::allocator<char> >&) !
! !
!----------------------------------------------------------------------------------!
like the option FROZEN_DISTANCE is not available anymore.
How can I make it to shorten the distance at each step using Cartesian coordinates?
Ouch, my bad - I haven’t seen that note at the end of the page - thanks!
Now the code works (no error) but it does not seem to change the distance between the two atoms I am specifying.
I.e., the “distance” variable changes but the RANGED_DISTANCE does not seem to set it correctly.
On a side note, I tried to run the same code but instead of single point energies I am requesting a constrained optimization at each step (‘energy’ changed to ‘optimize’) and I get the "List does not have 4*n entries as expected’. I think I get the origin of this message but what I am trying to do here is that at each step of the cycle I want the distance between the two atoms to be shortened, freeze the atoms in place and then do the job (single-point energy or full optimization).
What I would expect in this iterative job is that after each cycle the previous geometry is corrected according to the new set RANGE_DISTANCE but it does not seem to do so - am I missing something here or maybe the string is not correct?
Again, the ‘set optking’ command does not do anything but set the value of keywords. These two seem to be in conflict. Your frozen_cartesian prevents the motion of the O and C atoms. However the function of the ‘ranged_distance’ keyword is to shift the value of a coordinate during an optimization to the specified value.
So perhaps what you want can be achieved simply by omitting the frozen_cartesian keyword.
If you want to use optimizer machinery to construct the geometries before the optimization begins, it is possible to do this with the built-in interfragment coordinates, but the input is complex. For an energy scan (very similar to yours) but not through a TS see here:
Thanks for the answer! I have run additional tests and finally tried some approaches for the scan of around the rotation of the internal C-C bond in butane:
memory 16 gb
molecule butane {
0 1
C -1.8063 -0.7456 0.2148
C -0.2755 -0.6884 0.1961
H 0.1141 -0.9640 1.1867
C 0.2755 0.6885 -0.1961
H 1.3611 0.7054 -0.0222
H -0.1502 1.4510 0.4720
C -0.0150 1.0650 -1.6524
H 0.4269 0.3381 -2.3453
H 0.3924 2.0517 -1.9010
H -1.0944 1.0917 -1.8470
H 0.1114 -1.4442 -0.5025
H -2.2211 -0.0239 0.9294
H -2.1676 -1.7405 0.4994
H -2.2237 -0.5095 -0.7719
}
set {
basis 6-31G*
reference rhf # To ensure that the scf guess is right
g_convergence gau_loose # Sets the SCF convergence as scf=tight in G16, gau_loose for scans
dft_spherical_points 302 # The two following options makes the grid as 'finegrid' in Gaussian
dft_radial_points 75
dft_pruning_scheme robust # Generally safe and will speed things up
opt_type min # Options min, ts, irc
geom_maxiter 100
}
set optking dynamic_level 1
set optiking opt_coordinates cartesian
# Options for the scan
distance = -67.33
steps = 100
variation = 2.0
counter = 0
report = open("Scan_energies.txt","w") # To save the energy profile for check
while counter < steps:
counter = counter + 1
distance = distance + variation
distance_string = "1 2 4 7 %f %f" %(distance,distance) # Change the atom indexes as necessary
set optking ranged_dihedral = $distance_string
e = optimize('b3lyp')
butane.save_xyz_file('%d_opt.xyz' %counter,1)
report.write("Step: %d Distance: %f and Energy: %f\n" %(counter,distance,e))
report.flush()
report.close()
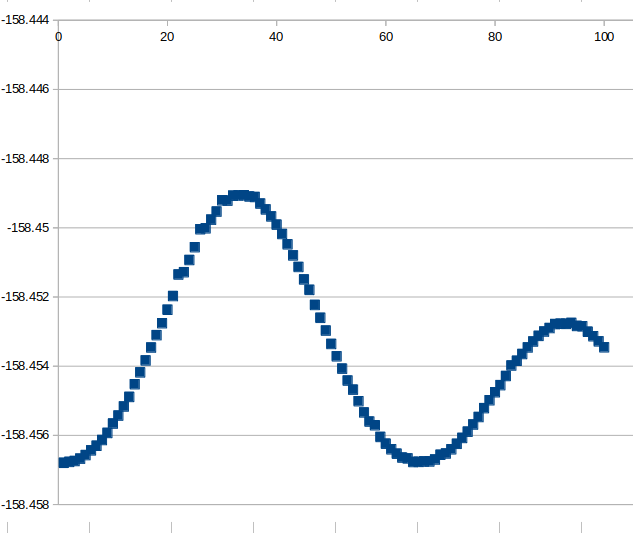
The calculation proceed well until the C-C-C-C dihedral is close to 0 degrees. Once there, the while cycle continues but it seems that PSI4 do not update the geometry anymore:
It is bizarre than once it reaches that geometry, although the “ranged_diedral” keyword is updated at each step, the geometry does not change and the optimization stop after the first SCF cycle even if not converged - see the ‘grep ‘~’ *out’ output below:
It looks like that when it reaches some type of geometries the system fails to continue.
I have tested the same on other type of rotations and even on a distance shortening calculation and I obtained the same behaviour.
I assume there is nothing helpful in the output or log files at this point?
Have you checked the gradients - do they look normal? I’m asking since a change in point group at zero could cause trouble with the electronic structure calculation. If the gradient is fine, then this looks like a bug to be fixed, and this fix is unlikely to happen soon. As a workaround on these types of scans (which do not in any event follow an IRC and are prone to discontinuities) you can try approaching the same point from the other direction.
Indeed, I was checking the log and this it seems that there is something weird in this block where it indicates that convergence was reached in one step but the Max Disp and RMS Disp are not:
2023-03-28:16:48:25,530 INFO [psi4.optking.convcheck:71] Performing convergence check.
2023-03-28:16:48:25,531 INFO [psi4.optking.convcheck:263]
==> Convergence Check <==
Measures of convergence in internal coordinates in au.
Criteria marked as inactive (o), active & met (*), and active & unmet ( ).
----------------------------------------------------------------------------------------------
Step Total Energy Delta E Max Force RMS Force Max Disp RMS Disp
----------------------------------------------------------------------------------------------
Convergence Criteria o 2.50e-03 * 1.70e-03 * 1.00e-02 * 6.70e-03 *
----------------------------------------------------------------------------------------------
1 -158.44909522 -1.58e+02 o 8.82e-06 * 2.63e-06 * 4.78e-01 1.02e-01 ~
----------------------------------------------------------------------------------------------
2023-03-28:16:48:25,531 INFO [psi4.optking.convcheck:193]
===> Final Convergence Report <===
----------------------------------------------------------------------------
| Required Criteria | One or More Criteria | Alternate Criteria |
----------------------------------------------------------------------------
| [x] max_force | | [x] flat_potential |
| [x] rms_force | | |
| [ ] max_disp | | |
| [ ] rms_disp | | |
----------------------------------------------------------------------------
2023-03-28:16:48:25,531 INFO [psi4.optking.stepAlgorithms:266] Convergence check returned True
2023-03-28:16:48:25,531 INFO [psi4.optking.optimize:286] Converged in 1 steps!
2023-03-28:16:48:25,531 INFO [psi4.optking.optimize:287] Final energy is -158.4490952242766
2023-03-28:16:48:25,531 INFO [psi4.optking.optimize:288] Final structure (Angstroms):
Fragment 1 (Ang)
C -1.1873901573 -1.1018659125 0.1992320027
C 0.1041108017 -0.5858491096 0.8488146543
H 0.0529726508 -0.1982621751 1.8743585021
C 0.9562967760 0.3998725006 -0.0116529516
H 1.8423555600 -0.1341768781 -0.3780792606
H 1.3360694635 1.2042945379 0.6306730001
C 0.1754664903 1.2793788251 -0.9980495657
H -0.2982122167 0.6877080164 -1.7891759654
H 0.8504732141 1.9944817655 -1.4825106767
H -0.6113593431 1.8575828203 -0.5004003252
H 0.6261043360 -1.5484449322 0.9183558716
H -1.9169442374 -0.2996128036 0.0428195966
H -1.6592940708 -1.8538222540 0.8423313761
H -0.9998229866 -1.5732974341 -0.7721808705
2023-03-28:16:48:25,532 INFO [psi4.optking.optimize:432] Optimization Finished
Hi! It looks like you’ve encountered an interesting edge case. flat_potential is normally a guard against continuing to optimize in flat regions of the PES. Because your convergence criteria are loose enough, the optimizer has gotten stuck at what it thinks is a stationary point. The forces at this particular point are low enough that despite the attempted step being large the optimizer is still quitting. The easy solution is to manually set the convergence criteria to the values for GAU_LOOSE
You may also try disabling the dynamic level. The scan quit for me at 15 steps after exceeding the dynamic level. Because the energy is increasing along the scan the optimizer classifies a large number of steps as “bad” and it eventually quit after exhausting its strategies to take more conservative downhill steps. The scan seems to perform well without the dynamic level on. I quit testing at point 54. Also, in case this was unintentional, note that your request for cartesian coordinates is not being recognized (it works fine without it though)