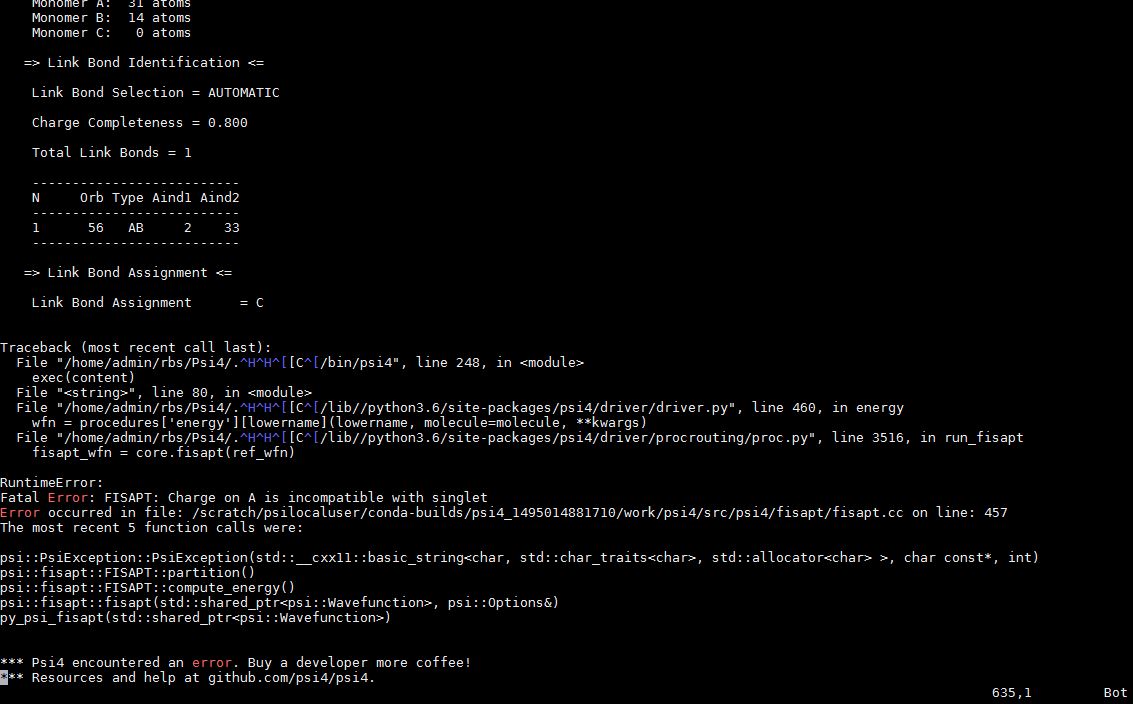
The partitioning of the molecule is closed shell because the SCF calculations don’t show any error. If my partition was not closed shell even the SCF calculations wouldn’t be done.
This error pops up only when the F-SAPT calculations start to run.
I have attached my input file below:
memory 1 GB
molecule mol {
0 1
O -2.09080353 -2.32271788 -0.13680149
H -0.98362848 -1.81070860 -0.74427008
O -3.44688855 -2.08959932 1.62287150
H 1.64508302 0.11815505 2.52135970
H -2.33364003 0.20000484 -2.31733416
C 1.00249418 0.90539504 2.11929114
H -4.48849883 -0.70001211 -0.86808663
H -1.86134690 1.86968967 -2.01677616
C -2.40233474 1.02610210 -1.60417397
C -2.88460909 -0.10399470 0.48054963
H -3.91635717 2.29278052 -0.74723701
C -2.77659945 -1.62998698 0.69163491
C -0.65811838 0.96648889 0.19542889
C -0.08603926 0.28990263 1.27339373
C -4.19960661 0.18322038 -0.29509699
N -1.83415048 0.57925989 -0.31902023
H -0.71458720 -0.41431133 1.80456187
H -2.90542298 0.36208143 1.46500599
H -4.49841719 1.35106295 -2.12994534
C -3.85325040 1.32652894 -1.25246634
H -5.01101186 0.41733955 0.39042567
H 0.52608713 1.36857422 2.99244469
C 0.11993320 2.07819516 -0.46195920
C 1.84345901 1.95332385 1.38594177
H 0.77380970 1.64729325 -1.22733846
C 0.95399462 2.87089169 0.55410428
H -0.55389409 2.76063238 -0.97569374
H 2.56474524 1.46378908 0.73041106
H 0.27732414 3.42711851 1.21056857
H 1.55392075 3.60832100 0.01681598
H 2.41838333 2.53378718 2.11093514
–
0 1
C 0.76605487 -1.35176510 -0.04638422
O -0.02116915 -1.45848109 -1.07766692
C 3.88978666 -0.00990930 -1.64791414
C 4.43866294 -0.87926637 0.53729422
H 5.86117432 0.03879013 -0.78770611
C 2.15909060 -0.97905251 -0.26651793
H 2.81310660 -1.74427871 1.63846621
H 1.83472208 -0.20323412 -2.24522330
H 0.55681420 -1.98869340 0.80933209
H 4.19610883 0.46251175 -2.57273917
H 5.16901259 -1.08465593 1.30953263
C 4.82785157 -0.25108394 -0.64377757
C 3.11248867 -1.24601846 0.72487957
C 2.56437813 -0.37551024 -1.46574611
symmetry c1
no_reorient
no_com
}
set {
basis jun-cc-pVDZ
scf_type df
guess sad
freeze_core true
}
energy(‘Fisapt0’)