Hello, Developers of PSI4 software:
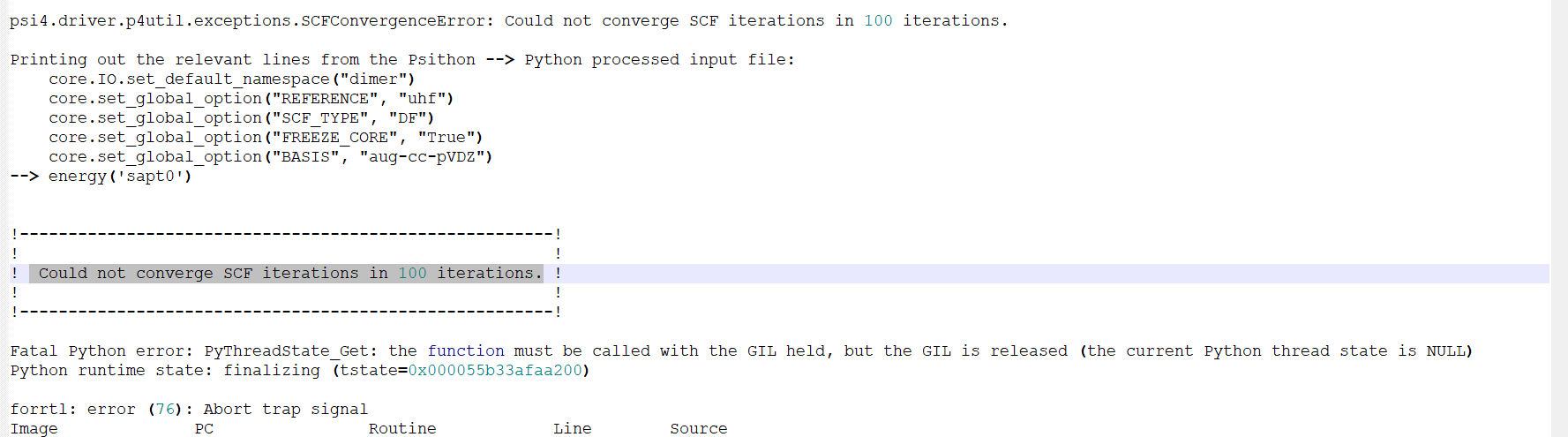
Calculation of the intermolecular interaction energy decomposition between the open and closed shell layers using SAPT0 results in the following error : Could not converge SCF iterations in 100 iterations. The structure has been optimizated in the Gaussian software without imaginary frequency.
we use the version of Psi4 is 1.6.1, here are my input and output file, I could not solve it, please help me, thanks very much.
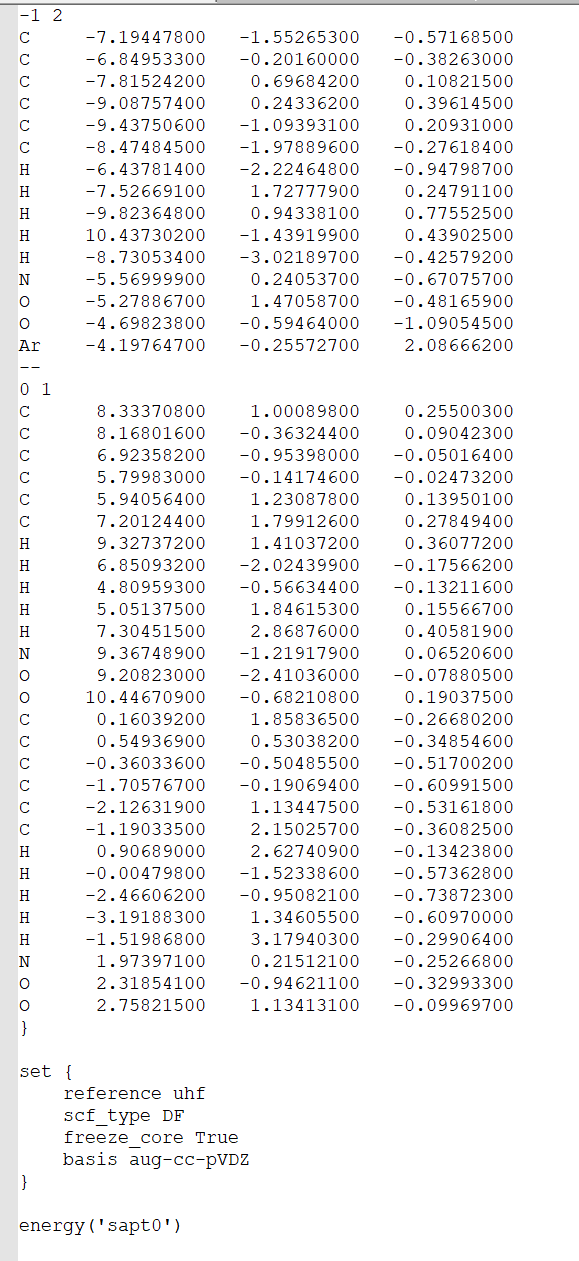
Hi, it is recommended to show the Cartesian coordinates data directly, so that someone can copy the Cartesian coordinates and check the geometry quickly. Showing a screenshot is not convenient for checking the geometry.
It seems that yourt input geometry is problematic/unreasonable: there is an H atom which is probably put in a wrong place. Besides, the current fragmentation is weird.
Thank you for your reminder, I have uploaded my input and output files, please help me to find out what the reason is! Thanks
NB3-2j-1-23.xyz (2.1 KB)
NB3-output.xyz (75.3 KB)
Please check the geometry of your molecule. The uploaded file confirmed the problem described in my previous reply.
Yeah, it is my carenessless. Let me try again. Thanks!