I’m using the SNS-MP2 plugin to calculate interaction between moleculers, but I have encountered difficulty when calculating interaction between [Li+] and H2O.
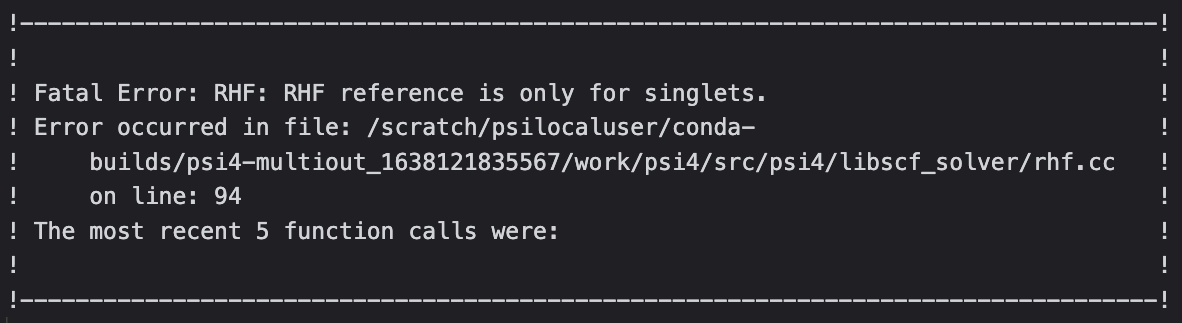
First, i choose RHF reference, it reports error as below:
Thanks for your reply. I’m so sorry i didn’t make myself clear.
The pair configuration in above input file is what I’m exactly interested in. I calculated Li-H2O just for a test. So I post above input file for help.
When I calculate Li-H2O or Li-C3H4O3, the program will report the same error. I don’t know whether the charge in system cause such a problem. This is what i want to figure out.
You can see an example here of how to specify the charge and multiplicity of a system with multiple fragments. That’s what 0 1 means. Charge 0, multiplicity 1. To specify a singlet cation, you need 1 1 for Li. A neutral singlet is 0 1, which is what is used by default. You don’t need to do anything for pyruvic acid(?).