Hi everyone, thanks for reading my topic! I want to run the TDDFT calculation with the TDSCF module for some large systems (i.e. perylene bisimide dimer, 80 atoms).
I found that the excited state’s results are quite different from the results from the Q-Chem. However, when I do the same calculation with small molecules (i.e. H2O2), I found that the result is totally the same.
I am wondering if I can change my settings to get the same results for large molecules. Could you help me correct my input file? Thanks!
The attachment files are my input files and output files for h2o2 (h2o2.py, h2o2.dat h2o2.py (951 Bytes) h2o2.dat (12.0 KB)
) and PBI dimer (pbi.py, pbi.dat pbi.dat (73.7 KB) pbi.py (4.0 KB)
).
Sorry, I forgot to mention that I change the scf_type to df for PBI dimer calculation. But I don’t think it will change the results too much.
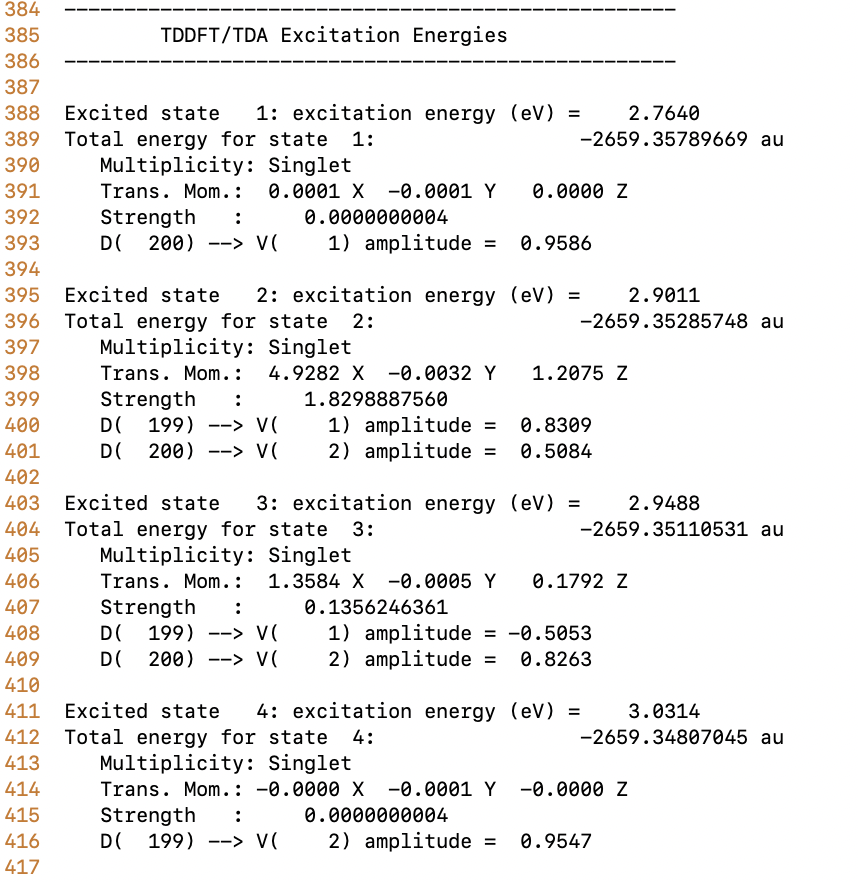
You can find that the PBI dimer results for the Psi4 and the Q-chem are quite different.
It is essential that I know which Psi4 version you’re using. We had an optimization for omega functionals. Between 1.4 and 1.5, we learned it was buggy, so we’ve temporarily disabled it until we can fix it. If you are using something lower than 1.5, upgrade immediately. This is likely the problem.
If upgrading does not fix your issue, could you attempt the same computation but for simpler DFT functionals and report back with whether they disagree with Q-Chem? If we’re seeing the same problem with TDHF, I will be looking at different parts of the code than if we only see this problem with omega functionals.
Thanks for your reply! Sorry about the late reply. I took a vacation recently and just came back to the office.
I used version 1.6a1.dev9 and 1.5. I tried both and these two versions gave the same result.
As you suggested, I tried TDHF and the result is the same with Q-Chem. As for the simpler DFT functionals, pbe result is also the same with Q-Chem.
However, several functionals (b3lyp, lrc-wpbe(h), wb97x-d3) give different result with Q-Chem. I tried to tighten the threshold but still can’t solve this issue.
Here I’d like to use one example to illustrate: PBI monomer (40 atoms) using lrc-wpbeh/6-31g*, the excitation energy difference between Psi4 and Q-Chem is about 2.5 eV. The main excitation is the same homo-lumo excitation.
Thanks for the report. This is troubling, and I’ll pass the information along to developers who know this part of the code better than I do.
Can you please give us the B3LYP files? That’s a relatively simple functional, and it’s easier for us to start investigating there. (The fact that PBE agrees but B3LYP does not tells me to look at the exchange hybrids.)
Holger and I have found and fixed the bug in range-separate TD-DFT. Expect a 1.5.1 release where this is fixed. Again, thank you for the report.
However, please give us the files you have demonstrating a bug with TD-B3LYP. There is not a chance that our bug fix will impact B3LYP, so if this bug is real, it’s still in the code.