It was my first time to use PSI4.
I want to use SPAT0/jun-cc-pvdz to decompose interaction energy, but the error faced to me in SCF not converged.
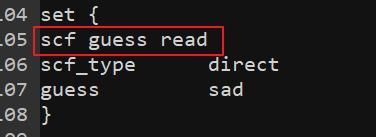
Now I am troubled by SCF not converged. So I want to use “guess read” to solove it. But I donnot know how to use it.
For example,
I have calculated the energy in other lower-level. I have had the fchk file.
But I donnot know how to use it.
For example, I just reaname the HF2.fchk to I-hf-jun-psi4-guess.chk ,then I submit the job
I-hf-jun-psi4-guess.inp with “scf guess read”. Is it ok to calculate successfully ?
Psi can write to FCHK files but cannot read from them at present.
Before guess read can be of any use, you’ll need to converge the SCF in Psi4. Can you post the output file, or is the information too sensitive? Without knowing the details of your computation, I can only give generic advice on SCF convergence in Psi.
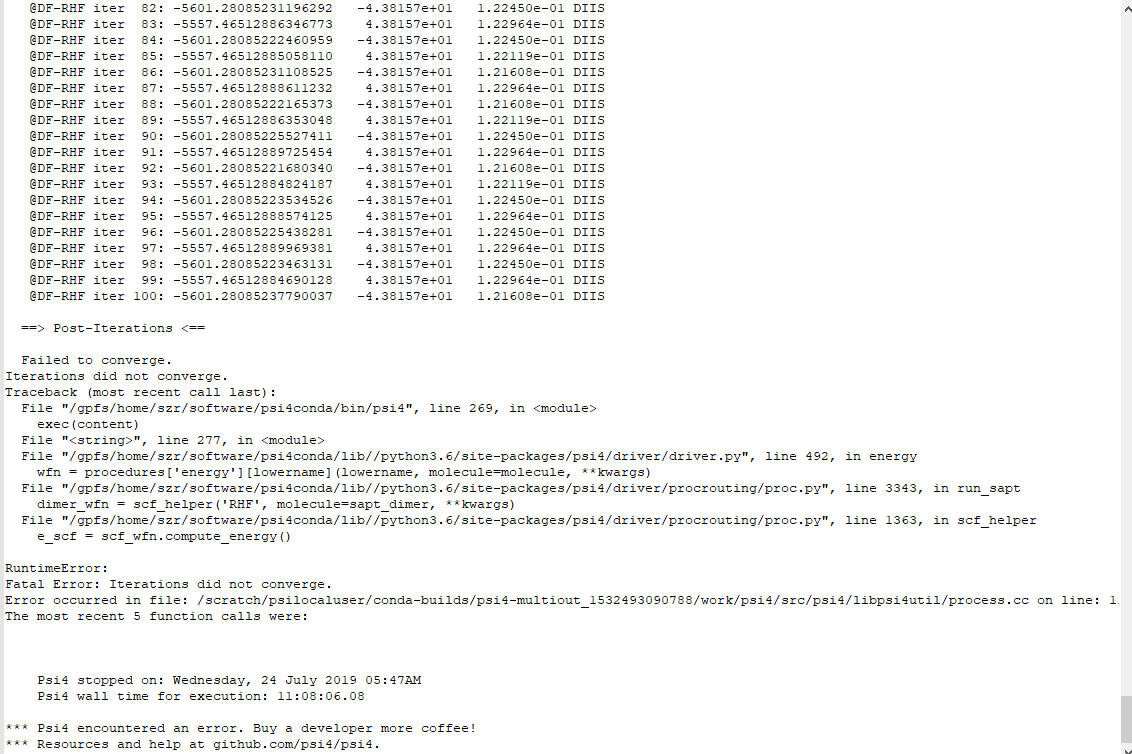
The large oscillations indicate that something is quite wrong.
Is this an unoptimized crystal structure? Correct units (bohr/Angstrom)?
Or a difficult electronic state?
I have tried other method to use “guess read” like this.
First I use cc-pvdz to calculate, then “guess read” the fchk to use the targated basis set (jun-cc-pvdz)
Yes, I use cc-pvdz without diffuse basis to calculate it. Then I use “guess read” to read the file with aug-cc-pvdz, I wish that it will help to SCF converge. Is it resonable to use it in the input file below.a-i-sapt0-guess-all.txt (13.2 KB)
Yes, I have updated the software in 1.3. And what can I do to solve the problem in SCF convergence? My jobs have 12 Iodine atoms, this is hard to converge in SCF.
This is a di-anion and as such expected to be unstable in the gas-phase!
Using a basis set without diffuse functions artificially traps the excess electrons, which may cause convergence of the SCF, but leads to an unphysical wave function.
If you require a SAPT decomposition you need to construct a model that is stable in the gas-phase.
In the future, please edit your previous post instead of making five posts in a row.
I’ll warn you that I’m not familiar with SAPT, so my advice will be limited to SCF convergence.
First, update your version of Psi4 and try the computation again. You’re using 1.2.1, but there were some important updates to SCF guesses in 1.3 that will make convergence easier. You can get the latest stable release here: https://admiring-tesla-08529a.netlify.com/installs/v132/
The problem of SCF converge is likely to be solved. The output file is here. But I have encontoured other question. The job usually is killed by itself. But there is no message left for me. I donnot know how to solve it. The output file has shown this problem. The job donnot report an error, but it finish with no result. And how to set the input file to output error files unitil it finished.a-i-sapt0-guess-def2svp.txt (274 KB)
My best guess is that 120 GB divided across 26 threads may not be enough memory for SAPT0 for a system with 2025 basis functions. I’ll check with one of the SAPT developers. Of course, I’d hope some kind of error message would get printed out. Did your cluster print out any kind of error file?
We have run SAPT computations of this size and larger in the past without issue and 120GB should be fine for memory. This is definitely worth looking into more.
@hokru where did that disk requirement come from? The size should be smaller than that as it only needs Qia density fitted tensors in disk.
@dgasmith The error i got points to disk problems, and that machine had only 740 GiB free space.
The exchange-dispersion part I mentioned did not finished (timer is zero)
...
Disp20 (OS) = -0.038042603435 [Eh]
Traceback (most recent call last):
File "/home/kruse/miniconda3/bin/psi4", line 289, in <module>
exec(content)
File "<string>", line 276, in <module>
File "/home/kruse/miniconda3/lib//python3.6/site-packages/psi4/driver/driver.py", line 561, in energy
wfn = procedures['energy'][lowername](lowername, molecule=molecule, **kwargs)
File "/home/kruse/miniconda3/lib//python3.6/site-packages/psi4/driver/procrouting/proc.py", line 3548, in run_sapt
e_sapt = core.sapt(dimer_wfn, monomerA_wfn, monomerB_wfn)
RuntimeError:
Fatal Error: PSIO Error
Error occurred in file: /scratch/psilocaluser/conda-builds/psi4-multiout_1562220055728/work/psi4/src/psi4/libpsio/error.cc on line: 128
The most recent 5 function calls were:
psi::PSIO::rw(unsigned long, char*, psi::psio_address, unsigned long, int)
I calculate it in my servers. But I donnot see any error file in my account, but it killed itself. And how to set in my input file ? then it will output the error files.
@Advanced When you run psi4 input.dat the output file will be a corresponding output.dat or you can add a -o /path/mycustomoutfile.out to control the name of the outfield generated.
I have calculated other molecules. And it is finished successfully. But how to divide the interaction energy? For example, Induction, dispersion and explusive terms can be summed into Van der Waals interaction. Is it correct ? And in closed-shell, the induction terms can be considered as polcarization effect and charge transfer effect, what do you think ?
Thank you~
Everybody, I have found that what made it. The memory is needed to be larger~ I use 370 G/ 20 threads to calculate it, then I got the result successfully. I am so happy. Gateful to all your help.
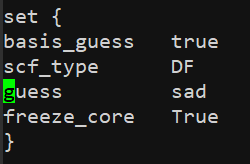
First , I faced the SCF convergence problem. I use def2-SVP basis set as the SCF guess to solve the problem.
Then I encountered the PSIO error. This is a memory error. You should increase the maximum memory to calculate it.
Thanks to all people.