Hi ,
I am performing geometry optimization for a set of alkynes (ethyne, propyne, 1-butyne, so on to 3-hexyne). I observed that atoms in linear angles are skipped in the list of internal coordinates printed in the output file.
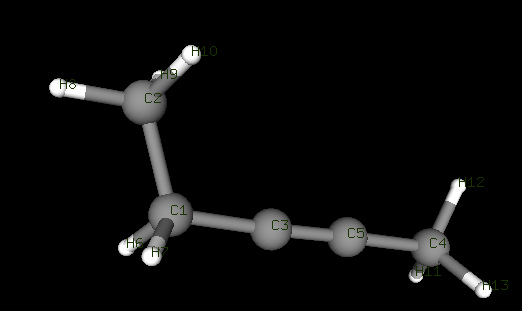
For example, consider the following molecule 2-pentyne, figure attached, with labelled atom numbers, where C1 indicates that the carbon atom has an index of 1. So the linear angles will be C1-C3-C5 and C3-C5-C4, while the corresponding dihedral shall be C1-C3-C5-C4 , indicating D(1,3,5,4)
The internal coordinates such as D(2,1,3,5) is replaced by D(2,1,4,11), so the atoms in the triple bonds are missing. I have only seen this for molecules with linear angles like alkynes, so far. Is there a way around to get this right?
Additional input info:
opttheory=“mp2”
optbasis=“6-31g(d)”
options={‘scf_type’: ‘df’, ‘g_convergence’:‘gau’,‘freeze_core’:‘true’}
psi4.set_options(options)
Thanks
Payal
Optking questions are a job for @Rollin_King
Is the optimization failing? That is, are the Cartesian gradients going to zero? If not, you could try
opt_coordinates = both
or
opt_coordinates = cartesian
I have not worked on this code in a long time, but I believe what is supposed to happen is that these degrees of freedom are covered by the addition of two complementary linear bending modes. Perhaps you see them in your list (the bending complements might be at the bottom of the list). It is difficult to maintain a meaningful dihedral value/force/displacement for a dihedral angle within which the bends are ~180 degrees.
Thanks for your response @Rollin_King !
The cartesian gradients are not going to zero. I tried opt_coordinates=cartesian before posting this question. Doing so, although that one linear dihedral gets included , their corresponding angles still skip the atom in the linear angle.
After talking to a few of my colleagues here, I understood that any internal coordinate with a linear angle is skipped in any QM package. Strangely however, that did not happen with terminal alkynes, but only internal alkynes. I also tested this with Gaussian package and it does the same.
My goal is to compare these QM internal coordinates with those in MM.
For now, I am just comparing the internal coordinates I can extract from the file, as this is perhaps not a “bug”, but correct me if I am wrong.
Thanks
We used to have a keyword in psi4 that would simply compute and print out all bond lengths, angles, etc… Someone else might know if we still have that and/or if it skips linear-like dihedrals.
In addition, I wonder if we have a python function at the ready that would compute all such values for a given geometry. Perhaps such a function might give you what you need.
Yes, that kind of a function shall be helpful.
Thanks
Payal
