Hello, I am new to using psi4 and SAPT calculations. I was looking at an example of a SAPT calculation of a water dimer (https://github.com/psi4/psi4/blob/master/samples/sapt3/input.dat) and noticed that there was a ghost atom. I was searching for information about the ghost atom, i.e., what it does, where to place it, and how it affects the calculation. As far as I understand it speeds up the calculation, right? I run a quick test with and without the ghost atom, and while the SAPT calculation with the ghost atom indeed was a little bit faster, I also realised that the interaction energies were slightly different (e.g. the dispersion term was different by 0.8 kJ/mol). Is there more documentation on the ghost atom (besides https://psicode.org/psi4manual/master/psithonmol.html#ghost-atoms) that explains how to use it (e.g. how to determine its coordinates), and how it affects the calculation? I guess that the calculation without the ghost atom is more precise, right?
The ghost atom in the example is to add mid-bond basis functions. This reduces basis set truncation effects at this point in space and can help to get better results.
(SAPT: Symmetry-Adapted Perturbation Theory)
The calculation with the ghost atom will run slower in general, because you have a large basis set.
Taking timings on a tiny example are borderline meaningless unless you take serious effort (run thousands calculations and be aware of general system load)
Ok, thank you, interesting. So adding the ghost atom makes the calculation in general slower and more accurate.
Do you have any reference that you can recommend to learn more about how to use ghost atoms?
ghost atoms are just a technical trick to place basis functions in space, here at the mid point between a binding vector.
This ‘trick’ is most prominently used in the calculation of the counterpoise correction to correct the BSSE ( basis set superposition error). You will find plenty of reading material in basic textbooks and internet search.
Ok, thanks again for your explanation.
Would the addition of ghost atoms be expected to minimise the SAPT results of a water dimer almost tenfold as below?
I have one SAPT water dimer system with a ghost atom and one without. Is the explanation only the ghost atom and basis set superpositon error? Would more ghost atoms increase the accuracy further?
# Define water dimer
#water_dimer = psi4.geometry("""
#O1
#H2 1 1.0
#H3 1 1.0 2 104.52
#x4 2 1.0 1 90.0 3 180.0
#--
#O5 2 """ + str(distances[i]) + """ 4 90.0 1 180.0
#H6 5 1.0 2 120.0 4 90.0
#H7 5 1.0 2 120.0 4 -90.0
#""")
water_dimer=psi4.geometry("""
O
H 1 B1
H 1 B2 2 A2
--
O 2 B3 1 A3 3 D3
H 4 B4 2 A4 1 D4
H 4 B5 2 A5 1 D5
B1 = 0.99025
B2 = 0.99025
A2 = 104.51302
B3 = """ + str(distances[i]) + """
A3 = 106.40712
D3 = 309.95750
B4 = 0.99025
A4 = 66.20205
D4 = 88.63853
B5 = 0.99025
A5 = 71.67858
D5 = 332.88215
""")
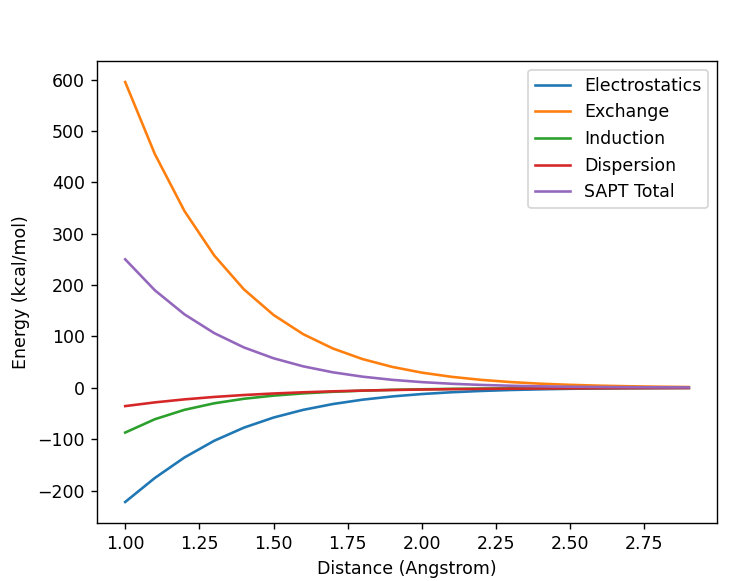
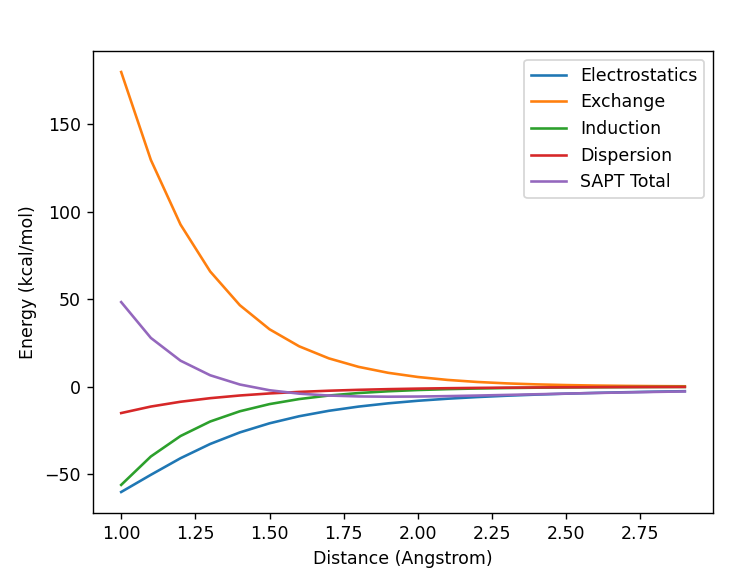
Thanks