Hi,
I’m interested in performing fractional electron (FE) calculations to assess the delocalization error of a double-hybrid density functional, B2PLYP-D3BJ/aug-cc-pVDZ. I’m using the frac module y’all developed (thank you!), calculating the SCF energy of water (fixed geometry) as a function of the number of electrons, N.
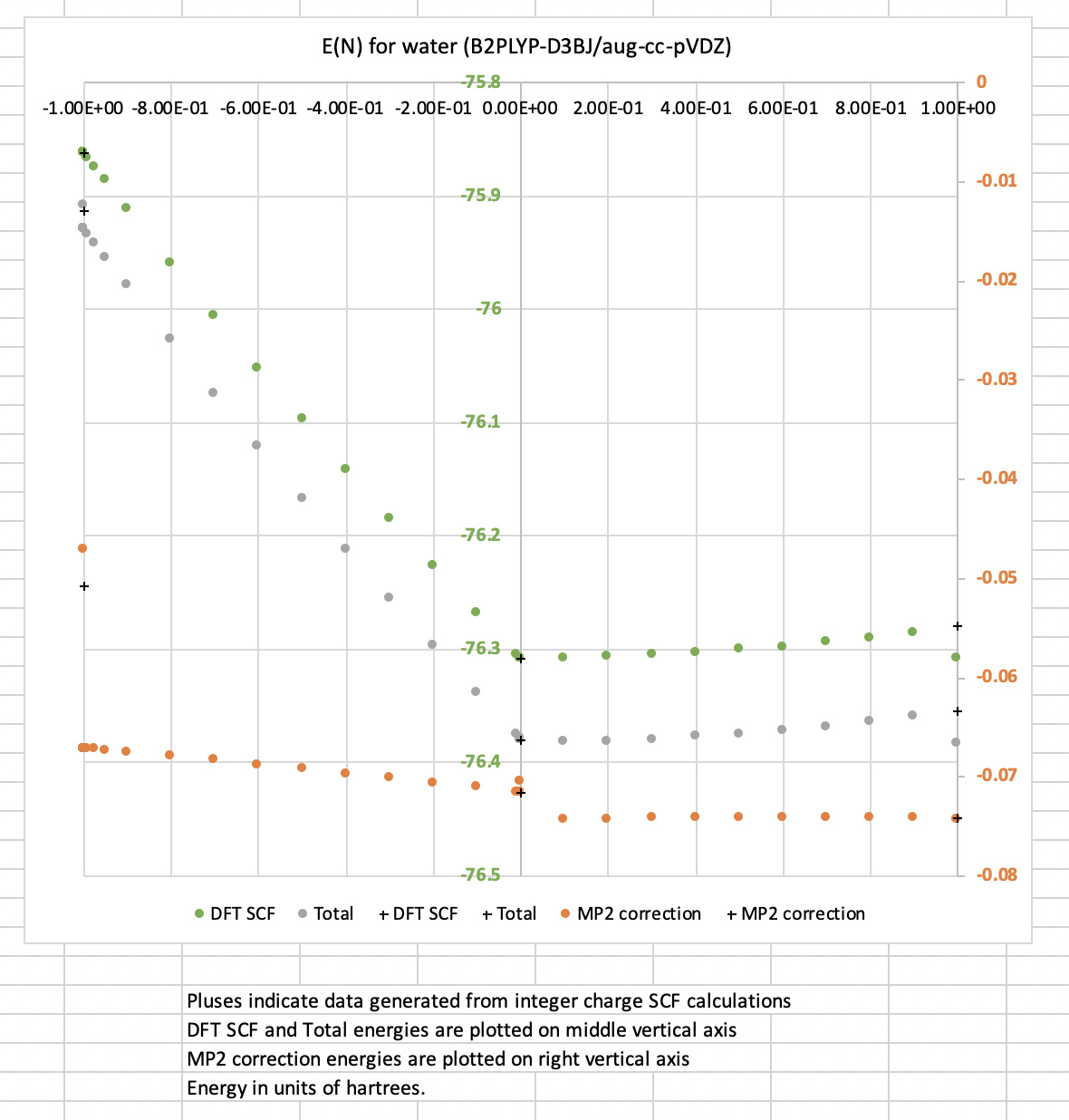
I’m having trouble reliably obtaining the endpoints of fractional scans with frac_traverse. For example, during a scan of HOMO occupancy, I can get three different energies (Hartrees) for the cation of water:
- FE-SCF extrapolation as
N--> -1:E~ -75.9288; - FE-SCF calculation with
N= -1:E= -75.9086; - Standard SCF calculation of doublet cation:
E= -75.9123.
I’ve attached the data and a visualization of the full E(N) curves at the bottom of this post. They show that in the above case, this discrepancy is caused by discontinuities in the MP2 correction at N=-1. However, in the anion case (N --> 1), discontinuities in the DFT contribution cause a similar problem.
Perhaps naively, I would expect all three measures of E(N) to be the same in both the anion and cation cases. Since I haven’t seen this behavior in the literature anywhere (e.g. DH-DFs are well behaved here), I was hoping y’all would be able to shed some light on this issue. As the deviation from linear FE-SCF energy as a function of N is a common measure of DE, it’s important to understand which endpoint I should be using in a linear interpolation.
Thanks in advance! Let me know if there’s any additional information I can provide to help clarify my question.
-Wes
Edit: I’m using Psi4 1.3.2, Git: Rev {HEAD} ecbda83. I’ve also attached input files that can be used to replicate the results. Thanks!
Plot:
Water_E_vs_N_plot
{kind=link}
Data:
Water_E_vs_N_data.txt (2.5 KB)
Psi4 inputs:
frac.dat (682 Bytes)
integer.dat (507 Bytes)