Dear Psi4 community,
I hope you’ll give me a hand.
I am studying non-covalent interactions, so there is a need to calculate the interaction energy. However, the dimer under study is linked by a pair of atoms to each other. To evaluate the energy of the contact, I decided to use the fi-sapt method. However, the results are rather strange. According to the QTAIM method, interaction energy = -2.1 kcal/mol. Calculated by the fi-sapt method, it turned out + 1.29 kcal/mol. That to recheck the results of fi-sapt I calculated sapt, for a dimer, replacing in one of the molecules a chlorine atom with hydrogen so that there was no contact of the chlorine 16 atom with oxygen 7. The interaction energy turned out to be -3.4 kcal/mol.
Please tell me why the calculated energy by the fi-sapt method turned out to be positive?
The calculated energy in a dimer with substituted chlorine for hydrogen can be considered the interaction energy of the contact Cl1—O22?
How do I calculate the interaction energy of contact Cl1 — O22?
Many thanks for your kind attention.
Best regards
Eugene
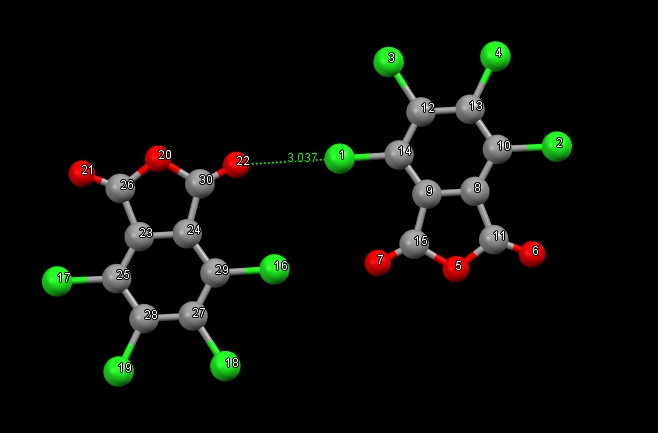
That’s the interaction energy between two molecules not between two functional groups or or atoms.
If you divide the value by two you have about the interaction energy of one Cl----O bond if you ignore the long range interaction between the whole molecules. Halogenated molecules like to behave unexpectedly compared to hydrogenated ones, so the long range interaction might not be ignorable.
QTAIM interaction estimation might or might not work, it’s an estimate and again, halogenated cases might need to be studied carefully before density-based relations are used.
F-SAPT0 will depend on your functional group and what bonds you cut. Cutting the C=O double bond is probably not a good idea. You can try doing C=O and Cl, or C=O and Cl-C fragments instead.
Not an expert on F-SAPT, others may have more hints.
Dear hokru,
thank you very much for answering the question
Best regards,
Eugene
I agree with hokru that cutting the C=O bond is a poor choice for this system. It is not recommended to use fragments that consist of only one atom, so I think using C=O and C-Cl is a good option.
Something else to consider is that the energy you want is probably the reduced analysis that includes the energy associated with the linking sigma bonds between the fragments. For the file you attached, the “F-SAPT energy” between the Cl1 and O fragments is +0.715 on line 137, not +1.292 on line 87.
Dear ccavender,
Thank you for explaining my results. Unfortunately, it is impossible to fragment the molecule at the Cl-C bond. The following error appears:
Exception: Orbital 58 is not complete over two link atoms. To avoid this error, please try to avoid cutting multiple bonds, aromatic rings, etc., in your definitions of fragments.
So I split it into two big pieces.
the first fragment is O = C-O-C = O and the second fragment is an aromatic ring. As a result of this fragmentation, I obtained interaction energy between fragments 1 and 2 equal to -1.143 kcal/mol.
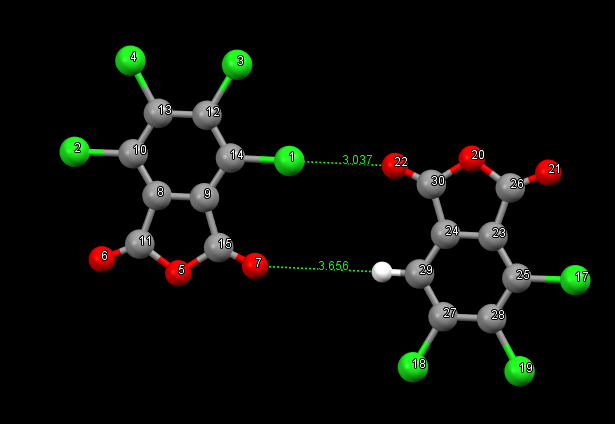
According to SAPT0 analysis, the interaction energy of a fragment in which one of the chlorine atoms is replaced by a hydrogen atom is estimated as -3.4 kcal/mol. It is assumed that the distances between oxygen and hydrogen atoms are larger than the Van der Waals radius, so there will be no contribution to the Cl-O interaction.
Please tell me how plausible my fi-sapt calculation results are with the new molecule fragmentation scheme?
Best regards,
Eugene
Hokru’s point about long-range interactions is going to be important here. You can see from your F-SAPT results that both of the Cl1 – O interactions sum to 2 * -1.143 kcal/mol = -2.286 kcal/mol, but the O – O interaction is -1.798 kcal/mol.
You could perhaps try an F-SAPT calculation with both Cl 1 and Cl 16 replaced by hydrogen so that you preserve the symmetry of the intermolecular interaction. The difference in total F-SAPT energy between that calculation and this calculation will be about the contribution of 2 Cl – O interactions plus the long-range interactions between the fragments. Then you can check whether the long-range interactions are similar between the two F-SAPT calculations to know how much of the difference in total F-SAPT energy comes from the Cl – O interactions.
Dear ccavender and hokru,
Thank you for the discussion. It helped me to understand the results.
Allow me to clarify a few questions.
Did I understand you correctly? To get the halogen bond energy, I need to get the difference between the total energy and the long-range energy (which is determined by the contribution of dipole-dipole interactions):
Eint(1)-Eint(3)=-4.4 +2.5 = -1.9 kcal/mol /2 = -0.95 kcal/mol energy halogen bond Cl-O
Apparently, this figure is very well consistent with the result of f-sapt -1.14 kcal/mol between fragments O = C-O-C = O and aromatic ring.
Can it be said that the dimer is mainly retained by halogen bonds and dipole-dipole interactions?
How to use f-sapt for similar molecules with iodine?
I also found the use of f-sapt in the detailed and interesting work 10.1021/acs.jpca.0c06266.

Best regards,
Eugene
The long-range energy is not just dipole-dipole interactions. It is any component of the noncovalent interaction that persists when the molecules are separated enough that the wavefunctions do not overlap significantly. The long-range contributions include electrostatic, induction, and dispersion energies. In contrast, short-range contributions are present only when wavefunctions overlap appreciably. The short-range contributions include exchange-repulsion, exchange-dispersion, and exchange-induction (which are all lumped into the SAPT “exchange” energy) and charge transfer (which is neglected in SAPT0). The long-range terms are still present at short range, so the “halogen bond energy” will include contributions from all of these terms.
It seems like the short-range exchange terms are negligible in complex 3, when the halogens are replaced by hydrogen. However, it is difficult to separate the long-range terms that arise specifically from the halogen bond from the long-range terms between other parts of the molecules. For example, there is clearly still a strong electrostatic interaction between the ground state electron densities of the two molecules in complex 3. The magnitude is likely not the same as the corresponding interaction in complex 1 because you’ve replaced an electon-withdrawing halogen with a hydrogen, and so the electron density in the aromatic ring is not the same between complex 1 and complex 3. The halogen atoms will also have longe-range interactions with the other parts of the molecule besides the accepting carbonyl group.
From your analysis, you can say to first order that each Cl contributes about 1 kcal/mol to the interaction energy of the complex. However, that interaction will include components other that the halogen bond with the carbonyl.
Dear ccavender,
Thanks for the detailed explanation!
Best regards,
Eugene
This topic was automatically closed 60 days after the last reply. New replies are no longer allowed.