Hi,guys, i am a new hand of using Psi4. I watched the video tutorial on Youtube recently.
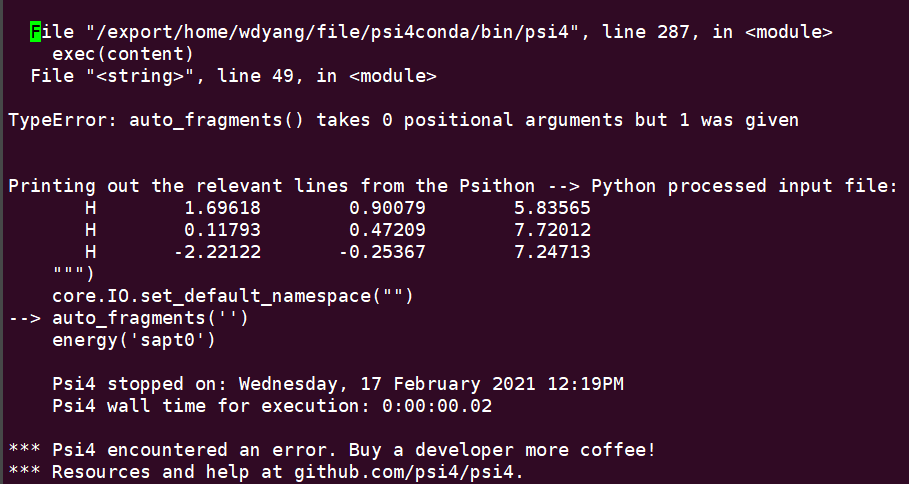
When I generate PSI input file from Avogadro accroding to the video(https://youtu.be/ou0dltm4zl4) and run program. It reminds error. Doesn’t auto_fragments(’’) function work?
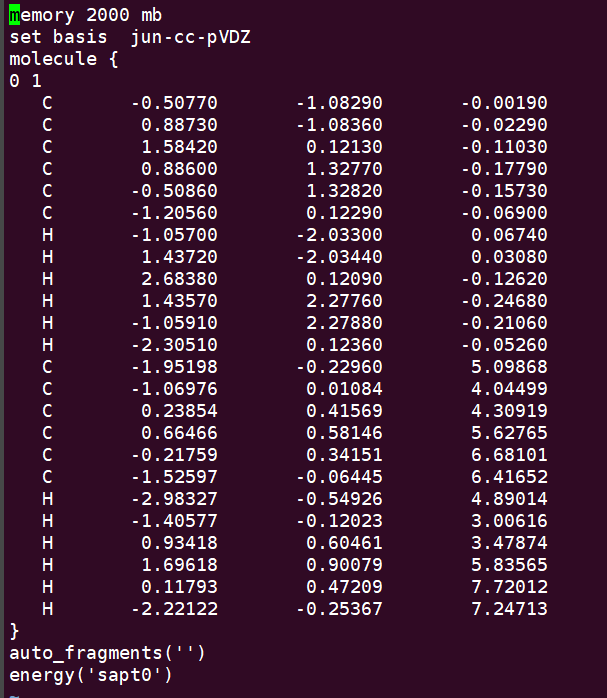
Input file:
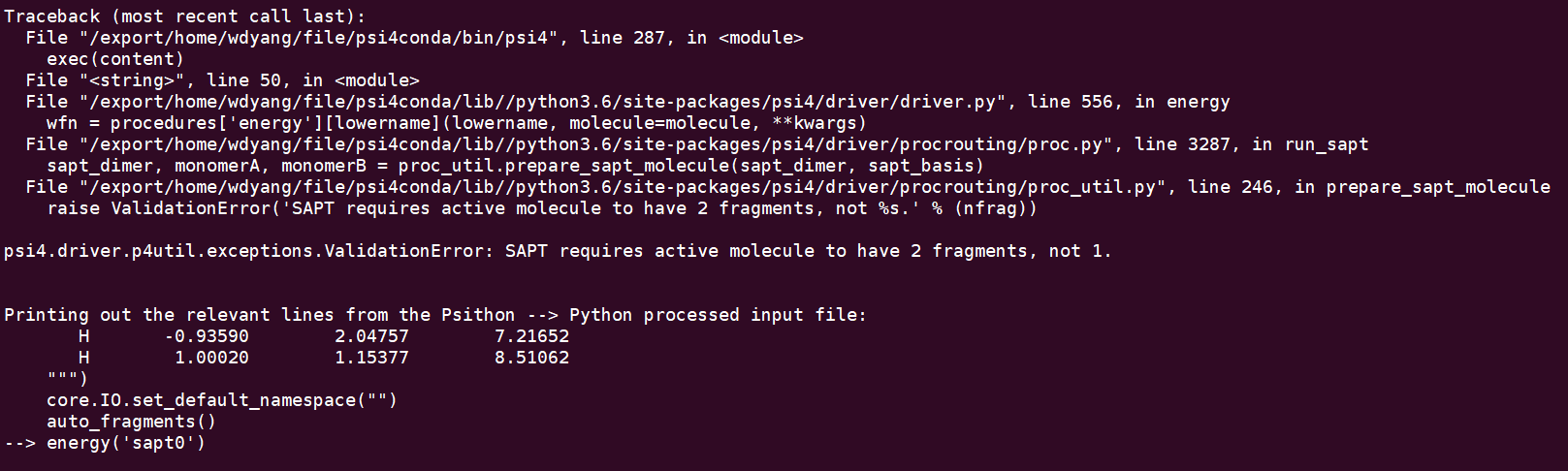
oh,thanks for your answer, i revise the input file according to your suggestion,
but it also doesn’t work. it remind me that i does not define 2 fragments.
of course, i can use the a line reading --to define 2 fragments
But if i doesn’t do this, can i use auto_fragments function to define? the video tutorial (https://youtu.be/ou0dltm4zl4) also use the input file from Avogadro and it works.