I’m new to psi4 and looking forward to using the python module within jupyter notebooks for class work.
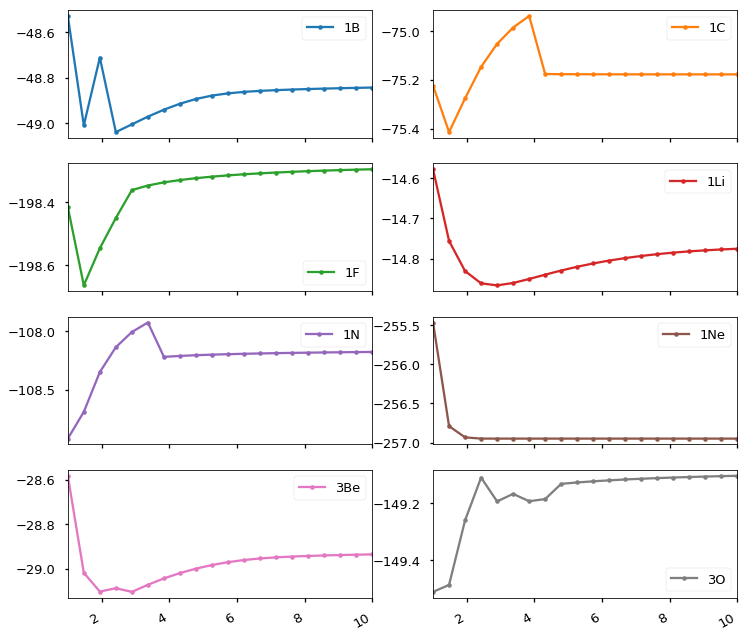
Following the PsiAPI Tutorial, tried to map potential curves of 2nd row diatomics. Certain diatomics give strange results despite converging without complaints. I’ve (naively) fiddled with reference wfs, setting charge and multiplicity explicitly or letting psi4 set these, changing e_convergence, basis sets, etc. and the unexpected (to me) results are reproducible. So I’m probably missing something obvious!
Environment: linux conda python 3.6, psi4 1.1+add49b9.
The legends give multiplicity and diatom’s element. All of these were rohf scf/6-31g* (similar results with rhf on singlets tho).
Specifically, these were generated by looping over r values with psi4.energy() and a template string:
r = np.linspace(1.25, 4, 20)
zmat = “”"
B
B 1 %s
“”"
The hackish Jupyter notebook is rendered on github.
Any suggestions or guidance?
Thanks again…the python module enables an exciting new work flow!
Steve
Bond breaking plots are best done from the outside, i.e. starting at a long distance. You then don’t have to worry about breaking the spatial symmetry of the orbitals. (I assume here you reuse the MOs from previous points)
Are you using molecular symmetry?
I have not much experience with ROHF, but I remember it being more prone to symmetry breaking than UHF.
I tried reversing the distances, working “outside-in”, and see the same results. I’m not (intentionally) re-using the orbitals. I’m passing a zmatrix to psi4.energy() for each value of r. I’m not doing an “update geometry”, but maybe I don’t understand what psi4 does under the hood after each energy calculation.
I wasn’t using molecular symmetry, either. The homonuclear diatomics were toy examples, leading eventually to heteronuclear systems. (The goal of all this is to see how MO energies vary in a period and fit to Morse potentials in an intro pchem course.)
I’m seeing similar results with rhf, rohf, and uhf. I’ve run similar calculations with nwchem and gaussian…those give smooth “Morse-like” curves, so I think I must be missing something in setting up the calculations for psi4.
Psi attempts to reuse orbitals from previous computations if they are available. Looking at it I would simply say that you are hitting different minima. Psi’s convergence for ROHF has, unfortunately, not been the best (for other guess id say we are quite good). You can try three things 1) use CUHF as the reference which helps with spin-contamination a bit and 2) set the following two options:
This doesnt seem to do great for molecules which is why its typically off, but usually helps out atom convergence. Finally, for 3), if you switch back to a UHF guess you can perform a stability analysis via: STABILITY_ANALYSIS FOLLOW option which will attempt to check if you are in a local minima and move the computation out the local minima and towards the global minima.
I tried dgasmith’s suggestions, but similar outcomes. I’ll play with different model chemistries/settings to work around what’s confusing psi4 (and me :).
Wow, this is embarrassing! Many quantum chemistry programs (including Psi4, apparently!) can have trouble setting appropriate initial guess orbitals for systems with high degrees of symmetry. I suspect that is what is happening here. One might think that simply turning off symmetry would solve this, but it does not — unless one deliberately attempts to scramble the symmetry of the guess orbitals (not necessarily a good idea…), the guess orbitals will still have symmetry even if run in “c1 symmetry”, and if those symmetries are wrong, it can be hard for the SCF procedure to break out of that local minimum solution into the correct global minimum solution (indeed, because the Hamiltonian commutes with the symmetry operations, it’s actually impossible to break out of the local minimum unless special steps are taken).
My old-school solution would be to manually specify the orbital occupations with the docc and socc keywords (with symmetry on) — these give the number of doubly occupied and singly occupied orbitals per irrep, respectively. Good exercise in undergraduate P-Chem to work out the correct values of those arrays.
However, ideally we developers should look at ways to more robustly get correct guess orbitals, or else be able to check if an irrep swap is called for during an SCF run (we used to do that in previous versions of Psi, it might have not made the jump to the latest version of the SCF code).
Ahh, this helps me understand what’s happening with the guess orbital symmetry vs molecular symmetry and some points in the earlier comments.
Having students work out docc/socc first is interesting. I think the typical assignment has them examine occupations post facto, but inverting the task makes for different problem…one that could give more insight than the “confirmatory” nature of the typical assignment. Or comparing the two to see what “gotchas” can arise when even simple systems aren’t simple. Cool.
At any rate, maybe a homonuclear diatomic series can become a test case someday :).