Hello, PSI4 communities,
I am new to PSI4, and I met some problems when I ran with PSI4 for my task of SAPT0. I guess maybe the frag 1 has the odd electrons but I changed the multi, it didn’t work.
Could someone please help me? A million thanks for your reply!!!
c: [0]
fc: [0.0]
fc: [0.0]
m: [1]
fm: [1]
fm: [1]
Traceback (most recent call last):
File “/public1/home/sc31713/miniconda/bin/psi4”, line 287, in
exec(content)
File “”, line 79, in
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/psi4/driver/molutil.py”, line 250, in geometry
geom, enable_qm=True, missing_enabled_return_qm=‘minimal’, enable_efp=True, missing_enabled_return_efp=‘none’)
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/qcelemental/molparse/from_string.py”, line 267, in from_string
**molinit,
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/qcelemental/molparse/from_arrays.py”, line 127, in from_input_arrays
verbose=1,
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/qcelemental/molparse/from_arrays.py”, line 386, in from_arrays
verbose=verbose,
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/qcelemental/molparse/chgmult.py”, line 501, in validate_and_fill_chgmult
c_final, fc_final, m_final, fm_final = reconcile(cgmp_exact_c, cgmp_exact_fc, cgmp_exact_m, cgmp_exact_fm)
File “/public1/home/sc31713/miniconda/lib//python3.7/site-packages/qcelemental/molparse/chgmult.py”, line 491, in reconcile
raise ValidationError(err)
qcelemental.exceptions.ValidationError: Inconsistent or unspecified chg/mult: sys chg: None, frag chg: [0.0, 0.0], sys mult: None, frag mult: [1, 1]
Printing out the relevant lines from the Psithon --> Python processed input file:
–
0 1
O 1.08106920 7.74959346 2.96835660
H 0.96944718 6.79014782 2.77644932
H 2.05118251 7.85615671 2.90916869
–> “”",“dimer”)
core.IO.set_default_namespace(“dimer”)
core.set_global_option(“SCF_TYPE”, “DF”)
core.set_global_option(“FREEZE_CORE”, “True”)
core.set_global_option(“BASIS”, “jun-cc-pVDZ”)
energy(‘sapt0’)
Psi4 stopped on: Wednesday, 10 June 2020 12:45PM
Psi4 wall time for execution: 0:00:00.69
*** Psi4 encountered an error. Buy a developer more coffee!
*** Resources and help at github.com/psi4/psi4
INPUT:
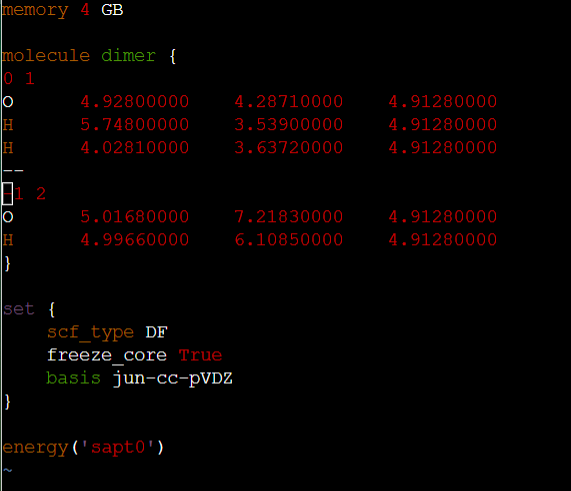
memory 4 GB
molecule dimer {
0 1
C -0.01397305 -0.06293349 -0.07866815
C 1.21924194 0.65302491 -0.04774983
C -1.24477669 2.06721817 -0.07135035
C -0.01869935 2.78290951 0.03306771
C -2.47256211 4.19456862 -0.07482150
C -1.24164965 4.89952407 0.02753957
C -3.70166040 6.32505011 -0.08147872
C -2.46505159 7.03468548 -0.05625220
C -4.93080994 8.45803708 -0.08316478
C -3.69692486 9.17007687 -0.09204825
C 2.45211931 -0.06305060 -0.08233281
C 3.68114055 0.64698716 -0.10845941
C 1.21929684 2.06396024 0.02336663
C 2.45718376 2.78264091 0.02715651
C -0.03339854 4.18703065 0.12665979
C 1.21950930 4.91583896 0.50246897
C -1.24308439 6.32975903 0.01047006
C -0.00305801 7.04152213 -0.00149754
C -2.46850096 8.46003591 -0.08988832
C -1.23848082 9.16814404 -0.12097128
C 4.91037328 -0.06377392 -0.14008652
C 6.14039746 0.64639465 -0.15954553
C 3.68278874 2.06686265 -0.07750529
C 4.91107074 2.77548799 -0.10703035
C 2.47093461 4.18606761 0.12145837
C 3.67928501 4.89836469 0.02300627
C 1.21820047 6.35543658 0.09342932
C 2.44044945 7.04098893 0.00209389
C -0.00925940 8.45984827 -0.10151252
C 1.21808585 9.16890931 -0.12440325
C 7.36866532 -0.06304566 -0.15985317
C 8.59727809 0.64652248 -0.15577637
C 6.14079117 2.06509956 -0.15239567
C 7.36903668 2.77504791 -0.15241711
C 4.91039843 4.19418607 -0.07834255
C 6.13926281 4.90546453 -0.10606459
C 3.68112345 6.32876217 0.01160774
C 4.90316452 7.03423446 -0.05486120
C 2.44623609 8.46035860 -0.09667362
C 3.67681289 9.16872688 -0.11479308
C 9.82754054 -0.06302504 -0.13411109
C 11.05719749 0.64739676 -0.10251790
C 8.59701903 2.06529934 -0.14903536
C 9.82700522 2.77573952 -0.10116827
C 7.36897524 4.19589646 -0.13067970
C 8.59874548 4.90566267 -0.10434830
C 6.13989559 6.32473736 -0.08133895
C 7.36914311 7.03361451 -0.08875228
C 4.90678264 8.46004605 -0.08539778
C 6.13543367 9.17022182 -0.08917896
O 1.23281230 5.01750277 2.00148262
H 1.11464133 4.10069635 2.34033261
0 1
O 1.08106920 7.74959346 2.96835660
H 0.96944718 6.79014782 2.77644932
H 2.05118251 7.85615671 2.90916869
}
set {
scf_type DF
freeze_core True
basis jun-cc-pVDZ
}
energy(‘sapt0’)
 {kind=link}