Dear friends, I hope you are fine.
I am trying to calculate the energy of a methane dimer at the CCSD(T)/aug-pVTZ level, but psi4 is giving me the error: “Segmentation fault (core dumped)”.
Below is my script:
memory 16gb
molecule ch4_dimer {
C 0.00000000 0.00000000 0.00000000
H 0.00000000 1.08452710 0.00000000
H -0.51125100 -0.36150900 0.88551270
H -0.51125100 -0.36150900 -0.88551270
H 1.02250190 -0.36150900 0.00000000
--
C 0.00000000 3.70000000 -0.00000000
H 0.00000000 2.61547290 -0.00000000
H -0.51125100 4.06150900 -0.88551270
H -0.51125100 4.06150900 0.88551270
H 1.02250190 4.06150900 0.00000000
}
set basis aug-cc-pVTZ
energy('ccsd(t)')
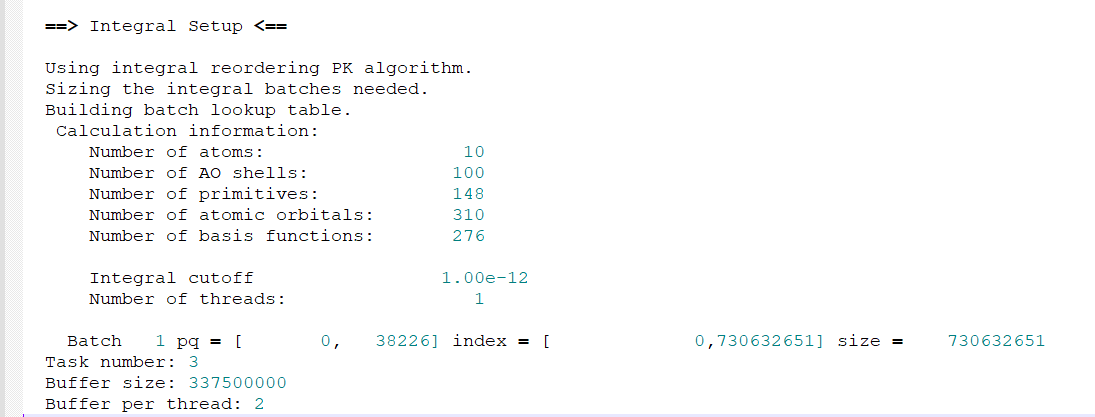
My output file ends here:
I get this error when I run this script on my pc, however on a cluster it is running normally.
I’m running this script with the following command: “psi4 -i test.dat -o test.out -n 1”
My version of psi4 is 1.1. and apparently I’m creating the SCRATCH directory correctly.
If you can kindly help me with this issue, I would be grateful. Thanks in advance.
1.1 is a very old version of Psi4. Without looking at any details, my immediate recommendation is to try out 1.5 and see if you still have the problem there.
Thanks for your fast reply, @jmisiewicz.
I think the problem was my version of psi4, the calculations in version 1.3 on the cluster I use didn’t give any errors.
Can you kindly help me with another related question? When I run the same methane dimer at the CCSD(T)/aug-pVQZ level I am getting the following error:
RuntimeError:
Fatal Error: PSIO Error
Error occurred in file: /scratch/psilocaluser/conda-builds/psi4-multiout_1557940846948/work/psi4/src/psi4/libpsio/error.cc on line: 128
The most recent 5 function calls were:
psi::PSIO::rw(unsigned long, char*, psi::psio_address, unsigned long, int)
psi::IntegralTransform::transform_tei_first_half(std::shared_ptrpsi::MOSpace, std::shared_ptrpsi::MOSpace)
I set the scratch directory where my script is with the command:
export PSI_SCRATCH=/home/users/emerson/my_directory
Doing this has a problem or is the error related to memory?
Below is my script:
memory 16gb
molecule ch4_dimer {
C 0.00000000 0.00000000 0.00000000
H 0.00000000 1.08452710 0.00000000
H -0.51125100 -0.36150900 0.88551270
H -0.51125100 -0.36150900 -0.88551270
H 1.02250190 -0.36150900 0.00000000
--
C 0.00000000 3.70000000 -0.00000000
H 0.00000000 2.61547290 -0.00000000
H -0.51125100 4.06150900 -0.88551270
H -0.51125100 4.06150900 0.88551270
H 1.02250190 4.06150900 0.00000000
}
set basis aug-cc-pVQZ
energy('ccsd(t)')
Below is my output:
teste1.txt (23.8 KB)
Thanks in advance for your time and help.
What gets written to stderr when you attempt this job?
Thanks for the reply, @jmisiewicz
The error file is filled with this message:
PSIO_ERROR: unit = 41, errval = 12
PSIO_ERROR: 12 (error writing to file)
Thanks in advance for your time and help.
That failure is occurring in the early part of the integral transformation and suggests that you’re running out of disk space.
@crawdad, many thanks for the answer!
Is there any way to know what disk space would be used in an estimated way?
In the directory I ran this script that gave an error there was ~170gb of space available.
Thanks in advance for your time and help.
Ignoring symmetry, the integral pre-sort requires (N*(N+1)/2)^2 double-precision words, which would be 145 GB for your calculation (for which N = 528 basis functions). It also requires you to hold the original integral file on disk, which would be another 9 GB. If there are other files taking up space on that disk, then you could be running out of room.