So I made the same script as you did but with import psi4 in a python script and I indeed miss something:
r = 1.0
psi4.geometry="""0 1
H
H 1 {0}""".format(r)
psi4.set_options({'basis': '3-21g',
'reference':'rhf',
'active': [1,0,0,0,0,1,0,0],
'restricted_docc':[0,0,0,0,0,0,0,0]})
Ecas, wfn = psi4.energy('casscf', return_wfn=True)
r = 1.1
psi4.geometry="""0 1
H
H 1 {0}""".format(r)
psi4.energy('casscf', ref_wfn=wfn)
gives the following error:
Traceback (most recent call last):
File "test_H2.py", line 14, in <module>
Ecas, wfn = psi4.energy('casscf', return_wfn=True)
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/lib/psi4/driver/driver.py", line 530, in energy
molecule.update_geometry()
AttributeError: 'NoneType' object has no attribute 'update_geometry'
So that’s why I don’t get different result: the geometry is certainly not updated, that’s all, when I use a restart wavefunction. I tried to get this update here but without any success, how should I proceed?
Also, I tried your script to orthogonalize the orbitals for the second geometry of H2 here, and it didn’t work straightforwardly. From this input:
molecule H2{
H
H 1 r
}
set {
reference rhf
basis 3-21g
active [1,0,0,0,0,1,0,0]
restricted_docc [0,0,0,0,0,0,0,0]
}
H2.r = 1.0
Ecas, wfn = energy('casscf', return_wfn=True)
print("CASSCF ORBITALS GEOMETRY 1")
print(wfn.Ca().nph)
H2.r = 1.1
Escf, wfnSCF = energy('scf', return_wfn=True)
print("SCF ORBITALS GEOMETRY 2")
print(wfnSCF.Ca().nph)
# import the script <== Change HERE
sys.path.insert(0, '/Users/bsenjean/Git_repositories/qchem_simulation/Notes/Embedding_project/AVAS')
from orthogonalize_orbitals import ortho_orbs
wfnSCF.Ca().copy(ortho_orbs(wfn,wfnSCF))
print("CASSCF ORBITALS FROM GEOMETRY 1 BUT ORTHOGONALIZED FOR GEOMETRY 2")
print(wfnSCF.Ca().nph)
Ecas2, wfn = energy('casscf', ref_wfn=wfnSCF, return_wfn=True)
print(Ecas)
print(Ecas2)
I get this output:
CASSCF ORBITALS GEOMETRY 1
(array([[ 0.37092414, 1.24482581],
[ 0.51019803, -0.9571024 ]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([[ 0.71712113, 1.15700169],
[ 0.91643852, -2.11514574]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64))
SCF ORBITALS GEOMETRY 2
(array([[ 0.33795149, 1.28231161],
[ 0.54922924, -0.94958934]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([[ 0.22654256, 1.31537109],
[ 1.50900811, -1.5438462 ]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64))
CASSCF ORBITALS FROM GEOMETRY 1 BUT ORTHOGONALIZED FOR GEOMETRY 2
(array([[ 0.37764796, 1.27118697],
[ 0.5194465 , -0.96620288]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64), array([[ 0.67716336, 1.15020539],
[ 0.86537484, -1.97780002]]), array([], shape=(0, 0), dtype=float64), array([], shape=(0, 0), dtype=float64))
invalid value encountered in less_equal
Traceback (most recent call last):
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/bin/psi4", line 331, in <module>
exec(content)
File "<string>", line 40, in <module>
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/lib/psi4/driver/driver.py", line 570, in energy
wfn = procedures['energy'][lowername](lowername, molecule=molecule, **kwargs)
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/lib/psi4/driver/procrouting/proc.py", line 4804, in run_detcas
ciwfn = mcscf.mcscf_solver(ref_wfn)
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/lib/psi4/driver/procrouting/mcscf/mcscf_solver.py", line 231, in mcscf_solver
total_step = diis_obj.extrapolate()
File "/Users/bsenjean/Git_repositories/psi4/objdir/stage/lib/psi4/driver/p4util/solvers.py", line 296, in extrapolate
invB.power(-1.0, 1.e-12)
RuntimeError:
Fatal Error: Matrix::power: C_DSYEV failed
Error occurred in file: /Users/bsenjean/Git_repositories/psi4/psi4/src/psi4/libmints/matrix.cc on line: 2280
The most recent 1 function calls were:
Printing out the relevant lines from the Psithon --> Python processed input file:
sys.path.insert(0, '/Users/bsenjean/Git_repositories/qchem_simulation/Notes/Embedding_project/AVAS')
from orthogonalize_orbitals import ortho_orbs
wfnSCF.Ca().copy(ortho_orbs(wfn,wfnSCF))
print("CASSCF ORBITALS FROM GEOMETRY 1 BUT ORTHOGONALIZED FOR GEOMETRY 2")
print(wfnSCF.Ca().nph)
--> Ecas2, wfn = energy('casscf', ref_wfn=wfnSCF, return_wfn=True)
print(Ecas)
print(Ecas2)
!----------------------------------------------------------------------------------!
! !
! Fatal Error: Matrix::power: C_DSYEV failed !
! Error occurred in file: !
! /Users/bsenjean/Git_repositories/psi4/psi4/src/psi4/libmints/matrix.cc on line: !
! 2280 !
! The most recent 1 function calls were: !
! !
!----------------------------------------------------------------------------------!
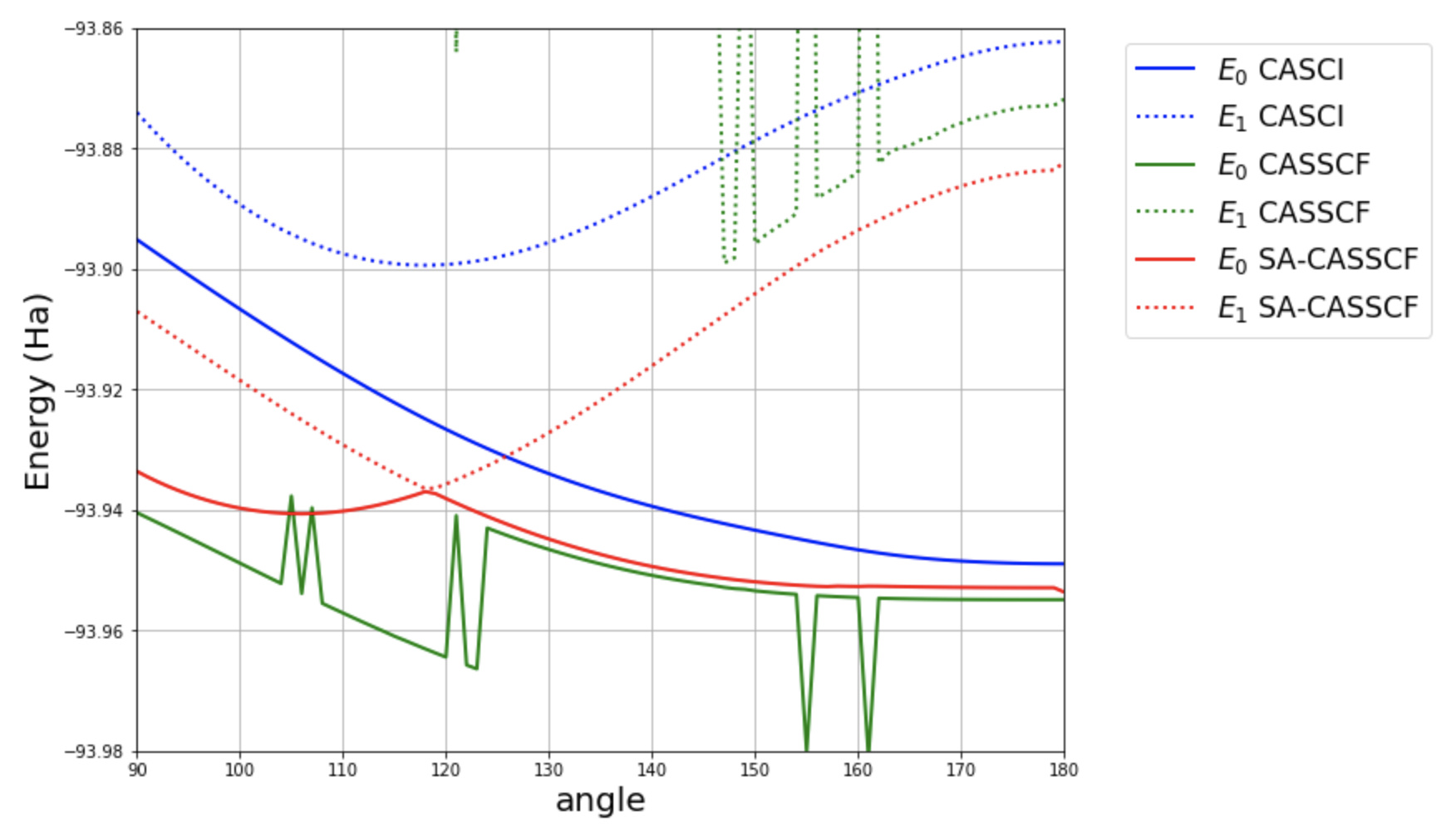


 . And we can see that the first iteration of MCSCF with the previous MCSCF orbitals is much closer to the converged MCSCF energy than with HF orbitals.
. And we can see that the first iteration of MCSCF with the previous MCSCF orbitals is much closer to the converged MCSCF energy than with HF orbitals.