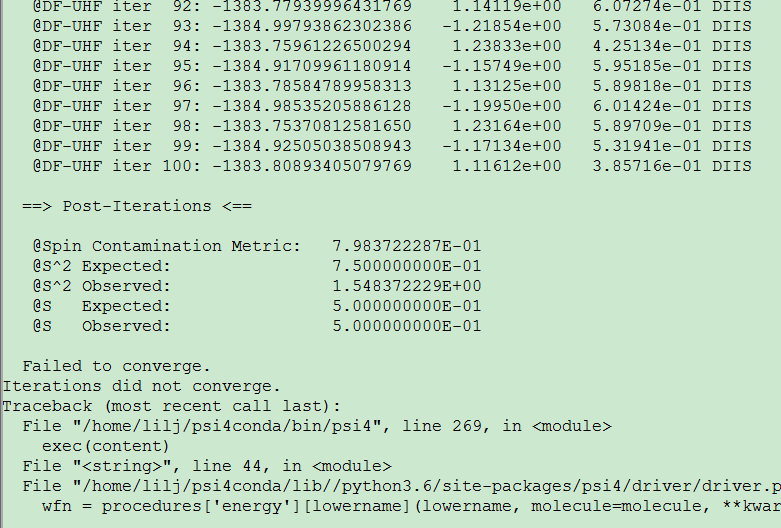
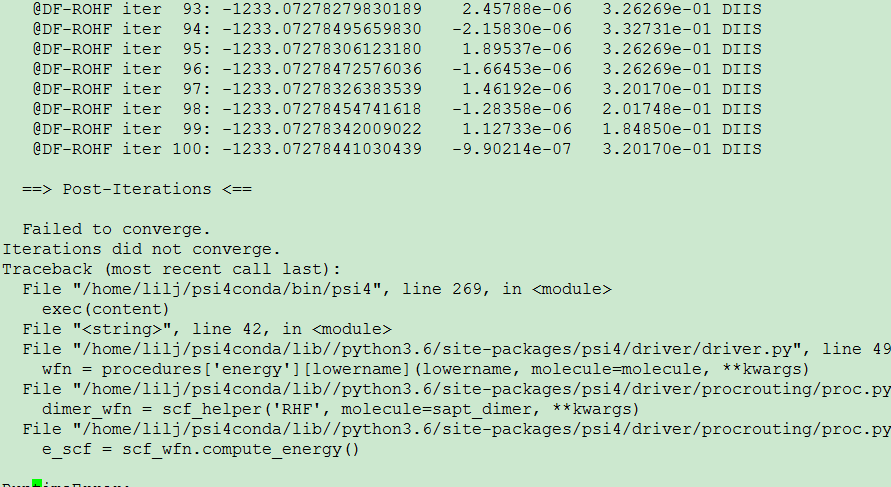
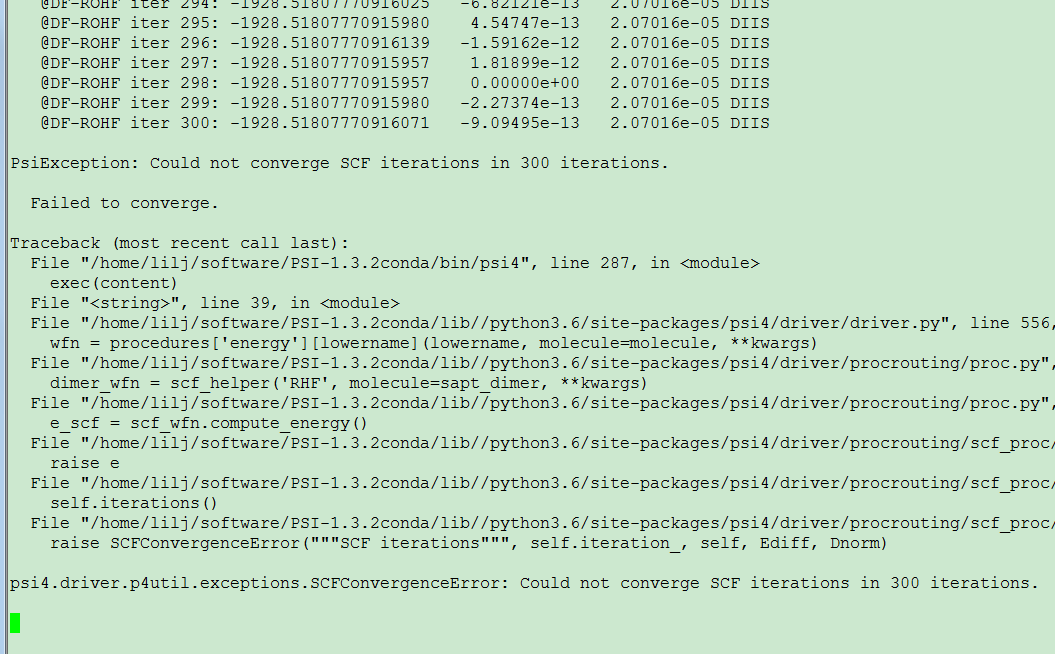
I made an energy decomposition analysis in the complex LnCl3H2O by sapt method. It works well for LaCl3H2O because there is no single electron in this conplex. But when it is applied to CeCl3H2O, a convergence problem occurs. The input file is shown below,
memory 40 GB
molecule dimer {
0 2
Ce -1.245901 -0.030094 0.049950
Cl -2.495563 2.194252 -0.517413
Cl 1.386678 -0.062614 0.280512
Cl -2.525909 -2.306411 -0.142389
--
0 1
O -0.902253 -0.347407 2.627758
H 0.046331 -0.409269 2.812856
H -1.359207 -0.969468 3.206986
}
dimerdist= 2.619900
set globals {
reference uhf
scf_type DF
basis def2-tzvp
df_basis_sapt def2-universal-jkfit
df_basis_scf def2-universal-jkfit
guess sad
}
energy('sapt0')
And the output is,
I tried to solve the problem by changing the reference into rohf. It does not work but seems closer to convergence.
My question is 1.how to solve the convergence failure? And 2.which reference should I select.
Thanks,
Best regards.
1 Like
We need more information to diagnose your SCF convergence failure. As we talk about here, we need to know what version of Psi you used. Some more recent versions of Psi have improved SCF convergence.
I don’t know whether UHF or ROHF are “better” for SAPT. You need one of the SAPT developers to answer that.
Thank you for reply. My version is 1.2. I will try a more recent version.
Please do! 1.3 has some improvements to our SCF guess.
Post back if that still doesn’t work.
I have updated my version to 1.3.2. But the problem still exists.
The input is,
memory 40 GB
molecule dimer {
0 2
Ce -1.24590100 -0.03009400 0.04995000
Cl -2.49556300 2.19425200 -0.51741300
Cl 1.38667800 -0.06261400 0.28051200
Cl -2.52590900 -2.30641100 -0.14238900
--
0 1
O -0.90880800 -0.34135400 2.57858400
H 0.03977600 -0.40321600 2.76368200
H -1.36576200 -0.96341500 3.15781200
}
dimerdist= 2.5699
set globals {
reference rohf
basis def2-tzvp
df_basis_sapt def2-universal-jkfit
maxiter 300
scf_type df
}
energy('sapt0')
E_disp = get_variable('SAPT DISP ENERGY') * psi_hartree2kcalmol
E_elst = get_variable('SAPT ELST ENERGY') * psi_hartree2kcalmol
E_exch = get_variable('SAPT EXCH ENERGY') * psi_hartree2kcalmol
E_ind = get_variable('SAPT IND ENERGY') * psi_hartree2kcalmol
E_tot = get_variable('SAPT TOTAL ENERGY') * psi_hartree2kcalmol
psi4.print_out("\n")
psi4.print_out(" Summary of SAPT result (kcal/mol)\n")
psi4.print_out(" Distance E_tot E_elst E_exch E_disp E_ind\n")
psi4.print_out("%s %6.3f %10.3f %10.3f %10.3f %10.3f %10.3f\n" % ("R=",dimerdist,E_tot,E_elst,E_exch,E_disp,E_ind))
I was able to mostly reproduce your results. This will take a while to run, but add the following keywords:
soscf true
soscf_max_iter 30
This uses a slower but more successful algorithm to converge the Hartree-Fock. soscf true uses the different algorithm, and soscf_max_iter controls how many “attempts” we give the iteration to work. Normally, the default value is fine, but this case is difficult enough that I found it needs a larger one. (For people who aren’t afraid of the technical details, we need to solve an equation by conjugate gradients for each step, and soscf_max_iter contols the number of iterations we give conjugate gradients to converge.)
If this continues to give you trouble, set soscf_print true and gives us the entire output file for debugging purposes.
I’ve added this molecule to our list of tricky SCF cases, so we can see if future improvements to the SCF code improve this.
1 Like
Thanks!
Following your advice, I went to have a test. And now I have good news and bad news. The good news is that the keywords work well to solve the convergence problem. However, the bad news is that they are only suitable to CeCl3H2O. When I applied them to PrCl3H2O with reference to be either rohf or uhf, the convergence problem happens again. Changing the soscf_max_iter value does not solve it. Now I set soscf_print true and upload the output files for you. Please take your time to analysis. And I will keep testing.
After downloading the output file, please change the extension from txt to rar and then uncompress it.
Appreciate your help!
output.txt (382.8 KB)
Thanks for the report.
Pr-uhf-40.out fails for a different reason. Psi’s SAPT requires that you can’t lower the energy by changing your orbitals, which we can usually interpret as “you have to be on the ground state”. It looks like you landed on some excited state. Add stability_analysis follow to instruct Psi to check if it can lower the energy any further, and if it can, to lower it.
Let us know if you have any other problems!